 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative Design of inorganic compounds using deep diffusion language models
Sep 30, 2023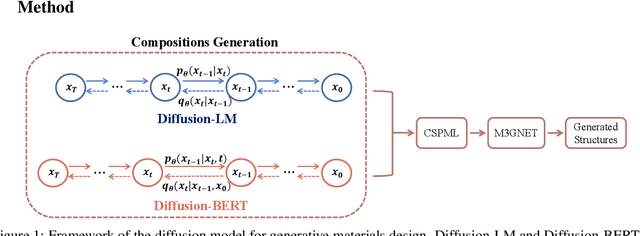
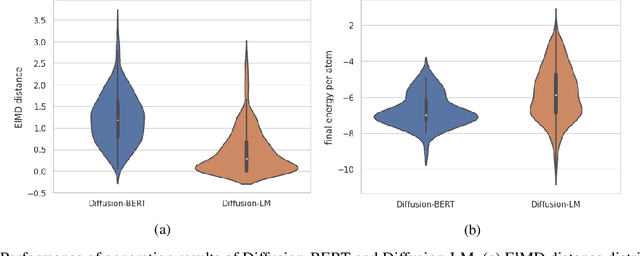
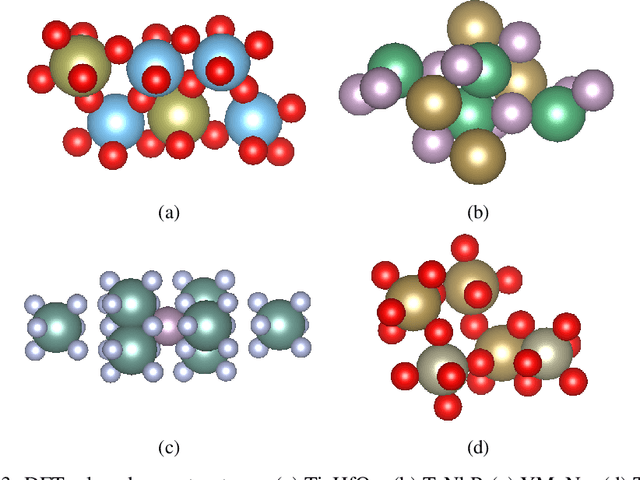
Due to the vast chemical space, discovering materials with a specific function is challenging. Chemical formulas are obligated to conform to a set of exacting criteria such as charge neutrality, balanced electronegativity, synthesizability, and mechanical stability. In response to this formidable task, we introduce a deep learning-based generative model for material composition and structure design by learning and exploiting explicit and implicit chemical knowledge. Our pipeline first uses deep diffusion language models as the generator of compositions and then applies a template-based crystal structure prediction algorithm to predict their corresponding structures, which is then followed by structure relaxation using a universal graph neural network-based potential. The density functional theory (DFT) calculations of the formation energies and energy-above-the-hull analysis are used to validate new structures generated through our pipeline. Based on the DFT calculation results, six new materials, including Ti2HfO5, TaNbP, YMoN2, TaReO4, HfTiO2, and HfMnO2, with formation energy less than zero have been found. Remarkably, among these, four materials, namely Ti2$HfO5, TaNbP, YMoN2, and TaReO4, exhibit an e-above-hull energy of less than 0.3 eV. These findings have proved the effectiveness of our approach.