 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMaterials Transformers Language Models for Generative Materials Design: a benchmark study
Jun 27, 2022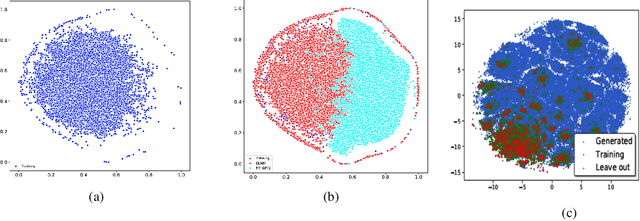
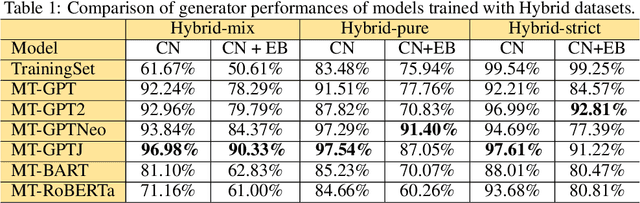
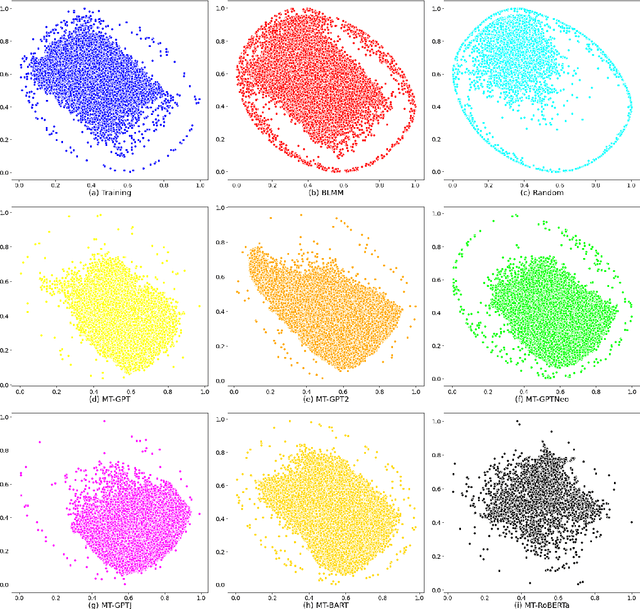
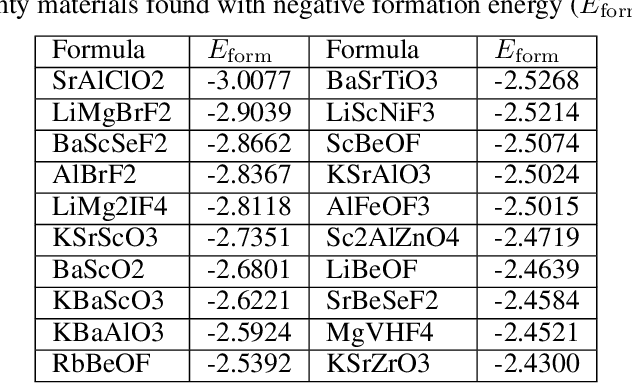
Pre-trained transformer language models on large unlabeled corpus have produced state-of-the-art results in natural language processing, organic molecule design, and protein sequence generation. However, no such models have been applied to learn the composition patterns of inorganic materials. Here we train a series of seven modern transformer language models (GPT, GPT-2, GPT-Neo, GPT-J, BLMM, BART, and RoBERTa) using the expanded formulas from material deposited in the ICSD, OQMD, and Materials Projects databases. Six different datasets with/out non-charge-neutral or balanced electronegativity samples are used to benchmark the performances and uncover the generation biases of modern transformer models for the generative design of materials compositions. Our extensive experiments showed that the causal language models based materials transformers can generate chemically valid materials compositions with as high as 97.54\% to be charge neutral and 91.40\% to be electronegativity balanced, which has more than 6 times higher enrichment compared to a baseline pseudo-random sampling algorithm. These models also demonstrate high novelty and their potential in new materials discovery has been proved by their capability to recover the leave-out materials. We also find that the properties of the generated samples can be tailored by training the models with selected training sets such as high-bandgap materials. Our experiments also showed that different models each have their own preference in terms of the properties of the generated samples and their running time complexity varies a lot. We have applied our materials transformer models to discover a set of new materials as validated using DFT calculations.
Physics Guided Generative Adversarial Networks for Generations of Crystal Materials with Symmetry Constraints
Mar 27, 2022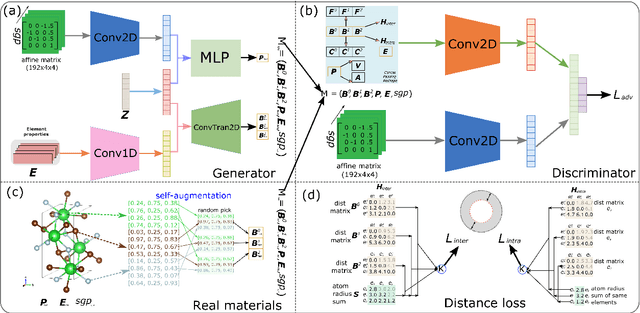
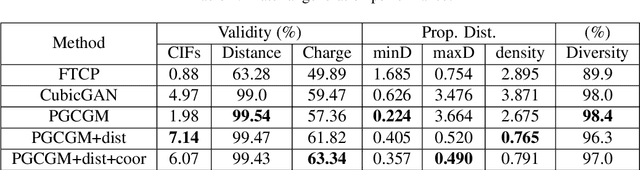

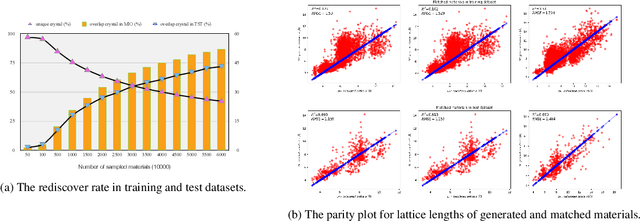
Discovering new materials is a long-standing challenging task that is critical to the progress of human society. Conventional approaches such as trial-and-error experiments and computational simulations are labor-intensive or costly with their success heavily depending on experts' heuristics. Recently deep generative models have been successfully proposed for materials generation by learning implicit knowledge from known materials datasets, with performance however limited by their confinement to a special material family or failing to incorporate physical rules into the model training process. Here we propose a Physics Guided Crystal Generative Model (PGCGM) for new materials generation, which captures and exploits the pairwise atomic distance constraints among neighbor atoms and symmetric geometric constraints. By augmenting the base atom sites of materials, our model can generates new materials of 20 space groups. With atom clustering and merging on generated crystal structures, our method increases the generator's validity by 8 times compared to one of the baselines and by 143\% compared to the previous CubicGAN along with its superiority in properties distribution and diversity. We further validated our generated candidates by Density Functional Theory (DFT) calculation, which successfully optimized/relaxed 1869 materials out of 2000, of which 39.6\% are with negative formation energy, indicating their stability.
Semi-supervised teacher-student deep neural network for materials discovery
Dec 12, 2021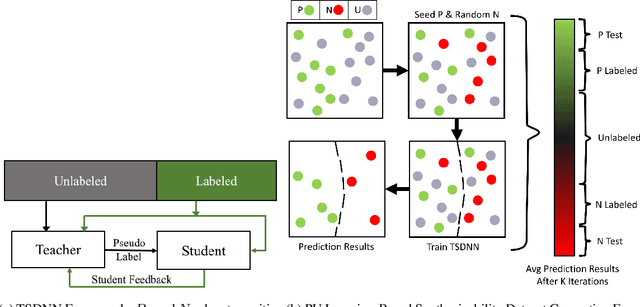
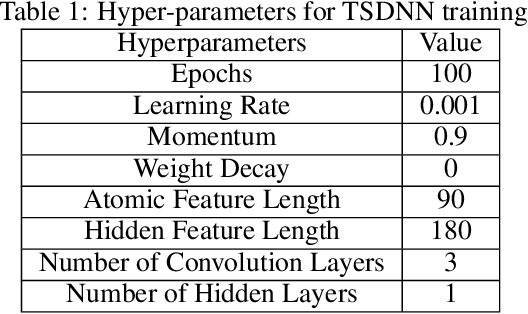
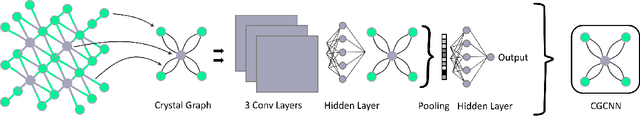
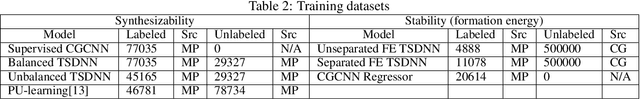
Data driven generative machine learning models have recently emerged as one of the most promising approaches for new materials discovery. While the generator models can generate millions of candidates, it is critical to train fast and accurate machine learning models to filter out stable, synthesizable materials with desired properties. However, such efforts to build supervised regression or classification screening models have been severely hindered by the lack of unstable or unsynthesizable samples, which usually are not collected and deposited in materials databases such as ICSD and Materials Project (MP). At the same time, there are a significant amount of unlabelled data available in these databases. Here we propose a semi-supervised deep neural network (TSDNN) model for high-performance formation energy and synthesizability prediction, which is achieved via its unique teacher-student dual network architecture and its effective exploitation of the large amount of unlabeled data. For formation energy based stability screening, our semi-supervised classifier achieves an absolute 10.3\% accuracy improvement compared to the baseline CGCNN regression model. For synthesizability prediction, our model significantly increases the baseline PU learning's true positive rate from 87.9\% to 97.9\% using 1/49 model parameters. To further prove the effectiveness of our models, we combined our TSDNN-energy and TSDNN-synthesizability models with our CubicGAN generator to discover novel stable cubic structures. Out of 1000 recommended candidate samples by our models, 512 of them have negative formation energies as validated by our DFT formation energy calculations. Our experimental results show that our semi-supervised deep neural networks can significantly improve the screening accuracy in large-scale generative materials design.
Crystal structure prediction of materials with high symmetry using differential evolution
Apr 20, 2021
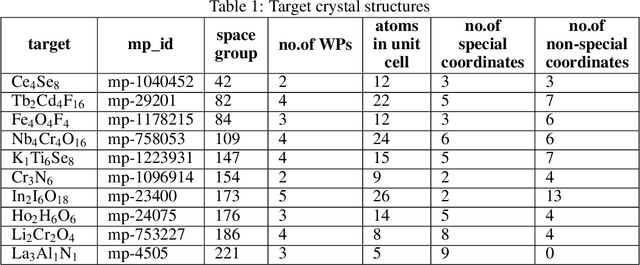
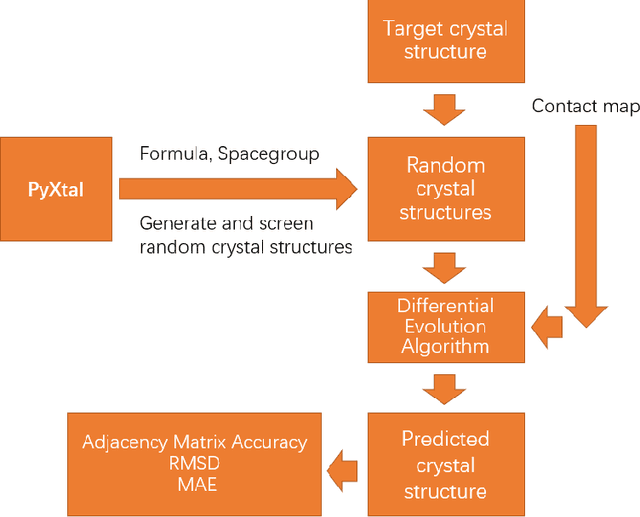
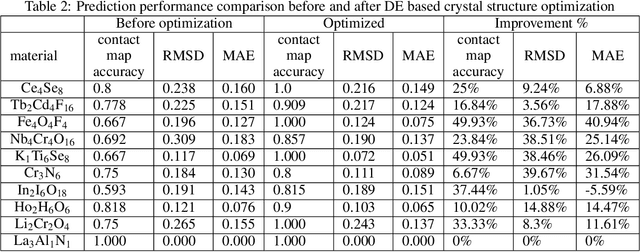
Crystal structure determines properties of materials. With the crystal structure of a chemical substance, many physical and chemical properties can be predicted by first-principles calculations or machine learning models. Since it is relatively easy to generate a hypothetical chemically valid formula, crystal structure prediction becomes an important method for discovering new materials. In our previous work, we proposed a contact map-based crystal structure prediction method, which uses global optimization algorithms such as genetic algorithms to maximize the match between the contact map of the predicted structure and the contact map of the real crystal structure to search for the coordinates at the Wyckoff Positions(WP). However, when predicting the crystal structure with high symmetry, we found that the global optimization algorithm has difficulty to find an effective combination of WPs that satisfies the chemical formula, which is mainly caused by the inconsistency between the dimensionality of the contact map of the predicted crystal structure and the dimensionality of the contact map of the target crystal structure. This makes it challenging to predict the crystal structures of high-symmetry crystals. In order to solve this problem, here we propose to use PyXtal to generate and filter random crystal structures with given symmetry constraints based on the information such as chemical formulas and space groups. With contact map as the optimization goal, we use differential evolution algorithms to search for non-special coordinates at the Wyckoff positions to realize the structure prediction of high-symmetry crystal materials. Our experimental results show that our proposed algorithm CMCrystalHS can effectively solve the problem of inconsistent contact map dimensions and predict the crystal structures with high symmetry.
Computational discovery of new 2D materials using deep learning generative models
Dec 16, 2020
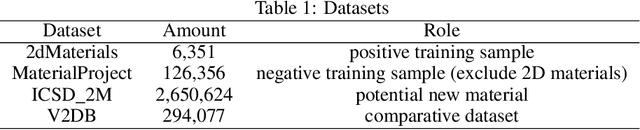
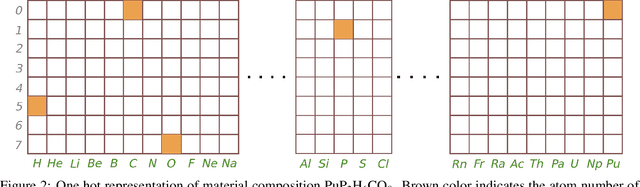
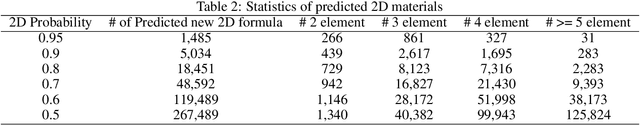
Two dimensional (2D) materials have emerged as promising functional materials with many applications such as semiconductors and photovoltaics because of their unique optoelectronic properties. While several thousand 2D materials have been screened in existing materials databases, discovering new 2D materials remains to be challenging. Herein we propose a deep learning generative model for composition generation combined with random forest based 2D materials classifier to discover new hypothetical 2D materials. Furthermore, a template based element substitution structure prediction approach is developed to predict the crystal structures of a subset of the newly predicted hypothetical formulas, which allows us to confirm their structure stability using DFT calculations. So far, we have discovered 267,489 new potential 2D materials compositions and confirmed twelve 2D/layered materials by DFT formation energy calculation. Our results show that generative machine learning models provide an effective way to explore the vast chemical design space for new 2D materials discovery.