 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePhysics Guided Generative Adversarial Networks for Generations of Crystal Materials with Symmetry Constraints
Paper and Code
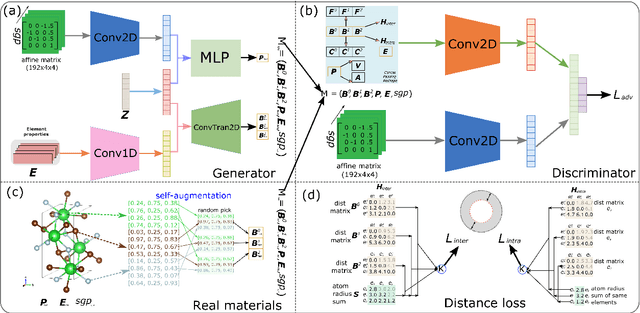
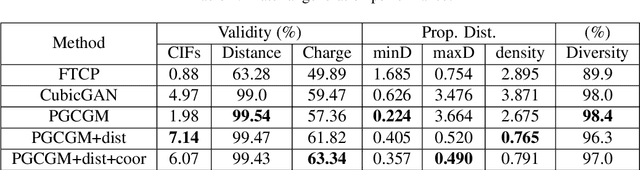

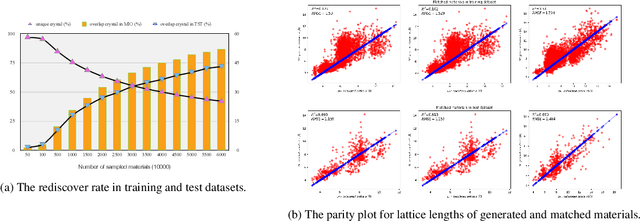
Discovering new materials is a long-standing challenging task that is critical to the progress of human society. Conventional approaches such as trial-and-error experiments and computational simulations are labor-intensive or costly with their success heavily depending on experts' heuristics. Recently deep generative models have been successfully proposed for materials generation by learning implicit knowledge from known materials datasets, with performance however limited by their confinement to a special material family or failing to incorporate physical rules into the model training process. Here we propose a Physics Guided Crystal Generative Model (PGCGM) for new materials generation, which captures and exploits the pairwise atomic distance constraints among neighbor atoms and symmetric geometric constraints. By augmenting the base atom sites of materials, our model can generates new materials of 20 space groups. With atom clustering and merging on generated crystal structures, our method increases the generator's validity by 8 times compared to one of the baselines and by 143\% compared to the previous CubicGAN along with its superiority in properties distribution and diversity. We further validated our generated candidates by Density Functional Theory (DFT) calculation, which successfully optimized/relaxed 1869 materials out of 2000, of which 39.6\% are with negative formation energy, indicating their stability.