 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Phylogenetic Approach to Genomic Language Modeling
Mar 04, 2025



Genomic language models (gLMs) have shown mostly modest success in identifying evolutionarily constrained elements in mammalian genomes. To address this issue, we introduce a novel framework for training gLMs that explicitly models nucleotide evolution on phylogenetic trees using multispecies whole-genome alignments. Our approach integrates an alignment into the loss function during training but does not require it for making predictions, thereby enhancing the model's applicability. We applied this framework to train PhyloGPN, a model that excels at predicting functionally disruptive variants from a single sequence alone and demonstrates strong transfer learning capabilities.
Genomic Language Models: Opportunities and Challenges
Jul 16, 2024



Large language models (LLMs) are having transformative impacts across a wide range of scientific fields, particularly in the biomedical sciences. Just as the goal of Natural Language Processing is to understand sequences of words, a major objective in biology is to understand biological sequences. Genomic Language Models (gLMs), which are LLMs trained on DNA sequences, have the potential to significantly advance our understanding of genomes and how DNA elements at various scales interact to give rise to complex functions. In this review, we showcase this potential by highlighting key applications of gLMs, including fitness prediction, sequence design, and transfer learning. Despite notable recent progress, however, developing effective and efficient gLMs presents numerous challenges, especially for species with large, complex genomes. We discuss major considerations for developing and evaluating gLMs.
Parallelizing Contextual Linear Bandits
May 21, 2021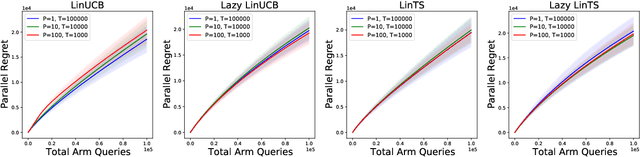
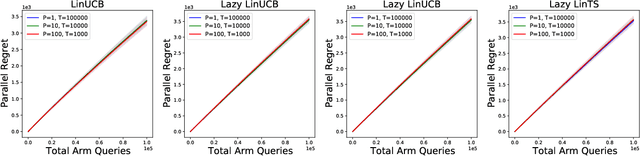
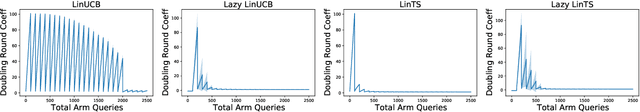
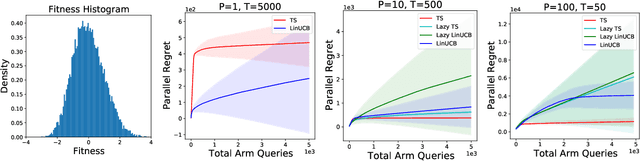
Standard approaches to decision-making under uncertainty focus on sequential exploration of the space of decisions. However, \textit{simultaneously} proposing a batch of decisions, which leverages available resources for parallel experimentation, has the potential to rapidly accelerate exploration. We present a family of (parallel) contextual linear bandit algorithms, whose regret is nearly identical to their perfectly sequential counterparts -- given access to the same total number of oracle queries -- up to a lower-order "burn-in" term that is dependent on the context-set geometry. We provide matching information-theoretic lower bounds on parallel regret performance to establish our algorithms are asymptotically optimal in the time horizon. Finally, we also present an empirical evaluation of these parallel algorithms in several domains, including materials discovery and biological sequence design problems, to demonstrate the utility of parallelized bandits in practical settings.
Evaluating Protein Transfer Learning with TAPE
Jun 19, 2019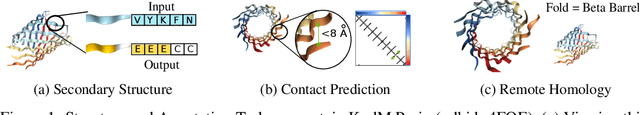
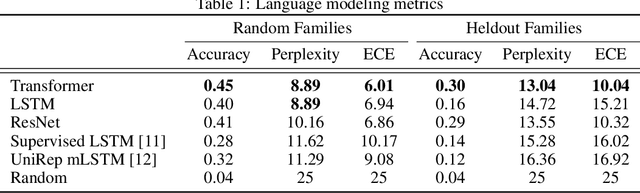
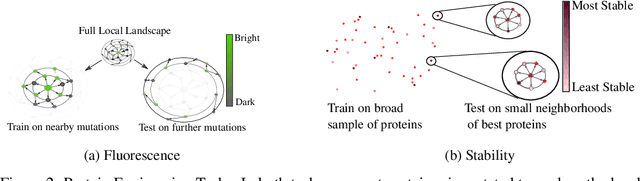
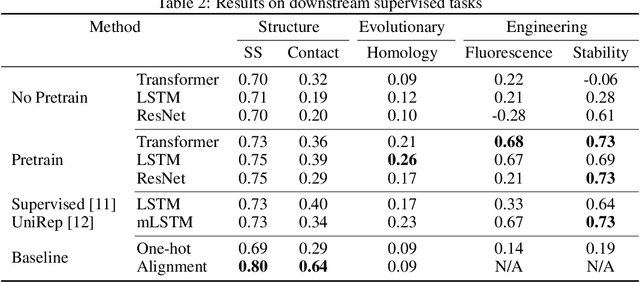
Protein modeling is an increasingly popular area of machine learning research. Semi-supervised learning has emerged as an important paradigm in protein modeling due to the high cost of acquiring supervised protein labels, but the current literature is fragmented when it comes to datasets and standardized evaluation techniques. To facilitate progress in this field, we introduce the Tasks Assessing Protein Embeddings (TAPE), a set of five biologically relevant semi-supervised learning tasks spread across different domains of protein biology. We curate tasks into specific training, validation, and test splits to ensure that each task tests biologically relevant generalization that transfers to real-life scenarios. We benchmark a range of approaches to semi-supervised protein representation learning, which span recent work as well as canonical sequence learning techniques. We find that self-supervised pretraining is helpful for almost all models on all tasks, more than doubling performance in some cases. Despite this increase, in several cases features learned by self-supervised pretraining still lag behind features extracted by state-of-the-art non-neural techniques. This gap in performance suggests a huge opportunity for innovative architecture design and improved modeling paradigms that better capture the signal in biological sequences. TAPE will help the machine learning community focus effort on scientifically relevant problems. Toward this end, all data and code used to run these experiments are available at https://github.com/songlab-cal/tape.
A Likelihood-Free Inference Framework for Population Genetic Data using Exchangeable Neural Networks
Nov 06, 2018

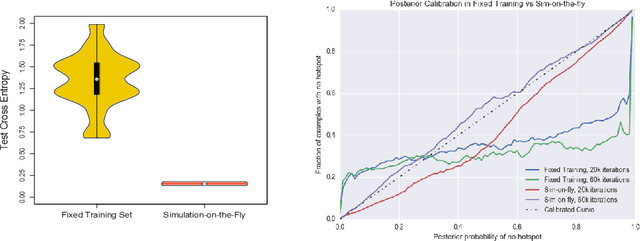
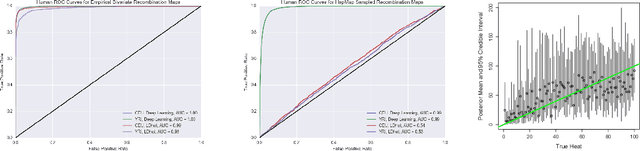
An explosion of high-throughput DNA sequencing in the past decade has led to a surge of interest in population-scale inference with whole-genome data. Recent work in population genetics has centered on designing inference methods for relatively simple model classes, and few scalable general-purpose inference techniques exist for more realistic, complex models. To achieve this, two inferential challenges need to be addressed: (1) population data are exchangeable, calling for methods that efficiently exploit the symmetries of the data, and (2) computing likelihoods is intractable as it requires integrating over a set of correlated, extremely high-dimensional latent variables. These challenges are traditionally tackled by likelihood-free methods that use scientific simulators to generate datasets and reduce them to hand-designed, permutation-invariant summary statistics, often leading to inaccurate inference. In this work, we develop an exchangeable neural network that performs summary statistic-free, likelihood-free inference. Our framework can be applied in a black-box fashion across a variety of simulation-based tasks, both within and outside biology. We demonstrate the power of our approach on the recombination hotspot testing problem, outperforming the state-of-the-art.
Tensor Decompositions via Two-Mode Higher-Order SVD (HOSVD)
Apr 19, 2017
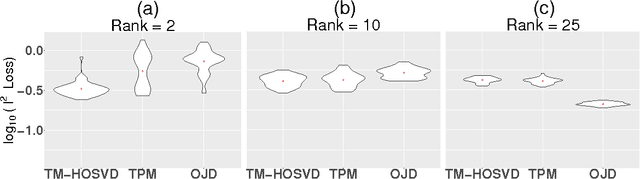
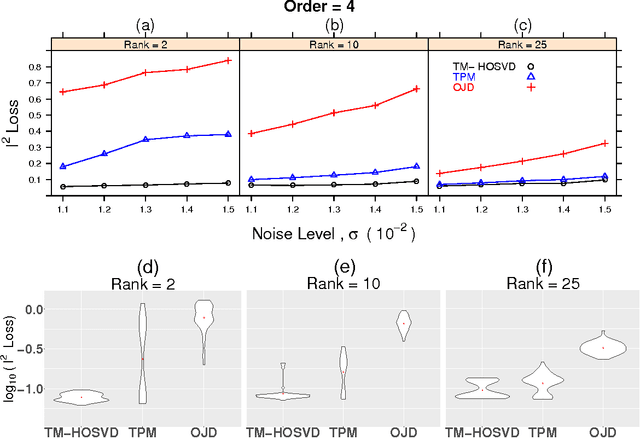
Tensor decompositions have rich applications in statistics and machine learning, and developing efficient, accurate algorithms for the problem has received much attention recently. Here, we present a new method built on Kruskal's uniqueness theorem to decompose symmetric, nearly orthogonally decomposable tensors. Unlike the classical higher-order singular value decomposition which unfolds a tensor along a single mode, we consider unfoldings along two modes and use rank-1 constraints to characterize the underlying components. This tensor decomposition method provably handles a greater level of noise compared to previous methods and achieves a high estimation accuracy. Numerical results demonstrate that our algorithm is robust to various noise distributions and that it performs especially favorably as the order increases.
* 33 pages, 5 figures