 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFlexible mean field variational inference using mixtures of non-overlapping exponential families
Oct 14, 2020

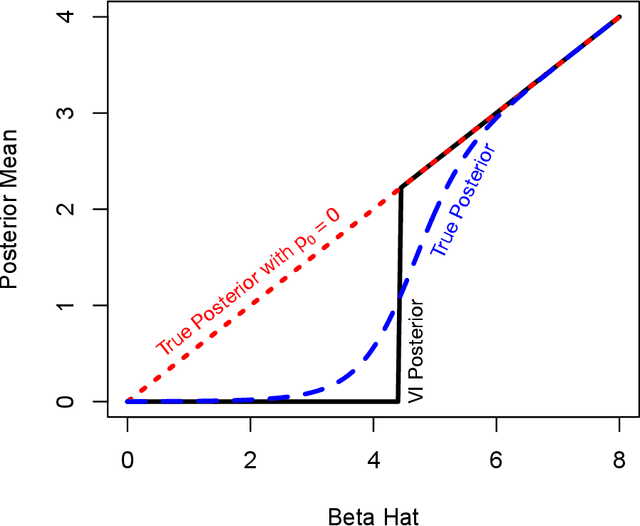

Sparse models are desirable for many applications across diverse domains as they can perform automatic variable selection, aid interpretability, and provide regularization. When fitting sparse models in a Bayesian framework, however, analytically obtaining a posterior distribution over the parameters of interest is intractable for all but the simplest cases. As a result practitioners must rely on either sampling algorithms such as Markov chain Monte Carlo or variational methods to obtain an approximate posterior. Mean field variational inference is a particularly simple and popular framework that is often amenable to analytically deriving closed-form parameter updates. When all distributions in the model are members of exponential families and are conditionally conjugate, optimization schemes can often be derived by hand. Yet, I show that using standard mean field variational inference can fail to produce sensible results for models with sparsity-inducing priors, such as the spike-and-slab. Fortunately, such pathological behavior can be remedied as I show that mixtures of exponential family distributions with non-overlapping support form an exponential family. In particular, any mixture of a diffuse exponential family and a point mass at zero to model sparsity forms an exponential family. Furthermore, specific choices of these distributions maintain conditional conjugacy. I use two applications to motivate these results: one from statistical genetics that has connections to generalized least squares with a spike-and-slab prior on the regression coefficients; and sparse probabilistic principal component analysis. The theoretical results presented here are broadly applicable beyond these two examples.
A Likelihood-Free Inference Framework for Population Genetic Data using Exchangeable Neural Networks
Nov 06, 2018

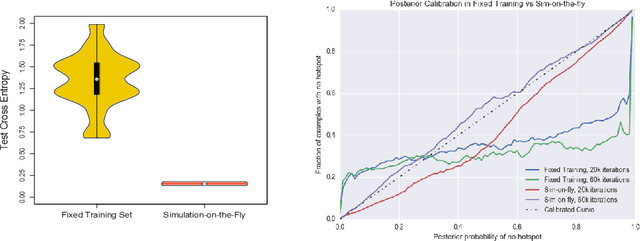
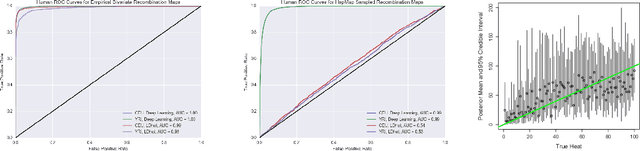
An explosion of high-throughput DNA sequencing in the past decade has led to a surge of interest in population-scale inference with whole-genome data. Recent work in population genetics has centered on designing inference methods for relatively simple model classes, and few scalable general-purpose inference techniques exist for more realistic, complex models. To achieve this, two inferential challenges need to be addressed: (1) population data are exchangeable, calling for methods that efficiently exploit the symmetries of the data, and (2) computing likelihoods is intractable as it requires integrating over a set of correlated, extremely high-dimensional latent variables. These challenges are traditionally tackled by likelihood-free methods that use scientific simulators to generate datasets and reduce them to hand-designed, permutation-invariant summary statistics, often leading to inaccurate inference. In this work, we develop an exchangeable neural network that performs summary statistic-free, likelihood-free inference. Our framework can be applied in a black-box fashion across a variety of simulation-based tasks, both within and outside biology. We demonstrate the power of our approach on the recombination hotspot testing problem, outperforming the state-of-the-art.