 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe Dance of Atoms-De Novo Protein Design with Diffusion Model
Apr 23, 2025



The de novo design of proteins refers to creating proteins with specific structures and functions that do not naturally exist. In recent years, the accumulation of high-quality protein structure and sequence data and technological advancements have paved the way for the successful application of generative artificial intelligence (AI) models in protein design. These models have surpassed traditional approaches that rely on fragments and bioinformatics. They have significantly enhanced the success rate of de novo protein design, and reduced experimental costs, leading to breakthroughs in the field. Among various generative AI models, diffusion models have yielded the most promising results in protein design. In the past two to three years, more than ten protein design models based on diffusion models have emerged. Among them, the representative model, RFDiffusion, has demonstrated success rates in 25 protein design tasks that far exceed those of traditional methods, and other AI-based approaches like RFjoint and hallucination. This review will systematically examine the application of diffusion models in generating protein backbones and sequences. We will explore the strengths and limitations of different models, summarize successful cases of protein design using diffusion models, and discuss future development directions.
Towards Cross-Modal Text-Molecule Retrieval with Better Modality Alignment
Oct 31, 2024Cross-modal text-molecule retrieval model aims to learn a shared feature space of the text and molecule modalities for accurate similarity calculation, which facilitates the rapid screening of molecules with specific properties and activities in drug design. However, previous works have two main defects. First, they are inadequate in capturing modality-shared features considering the significant gap between text sequences and molecule graphs. Second, they mainly rely on contrastive learning and adversarial training for cross-modality alignment, both of which mainly focus on the first-order similarity, ignoring the second-order similarity that can capture more structural information in the embedding space. To address these issues, we propose a novel cross-modal text-molecule retrieval model with two-fold improvements. Specifically, on the top of two modality-specific encoders, we stack a memory bank based feature projector that contain learnable memory vectors to extract modality-shared features better. More importantly, during the model training, we calculate four kinds of similarity distributions (text-to-text, text-to-molecule, molecule-to-molecule, and molecule-to-text similarity distributions) for each instance, and then minimize the distance between these similarity distributions (namely second-order similarity losses) to enhance cross-modal alignment. Experimental results and analysis strongly demonstrate the effectiveness of our model. Particularly, our model achieves SOTA performance, outperforming the previously-reported best result by 6.4%.
Graph Neural Networks for Double-Strand DNA Breaks Prediction
Jan 04, 2022
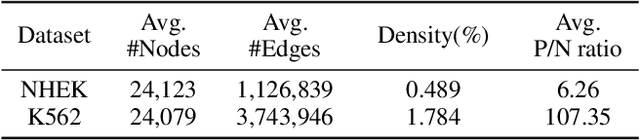

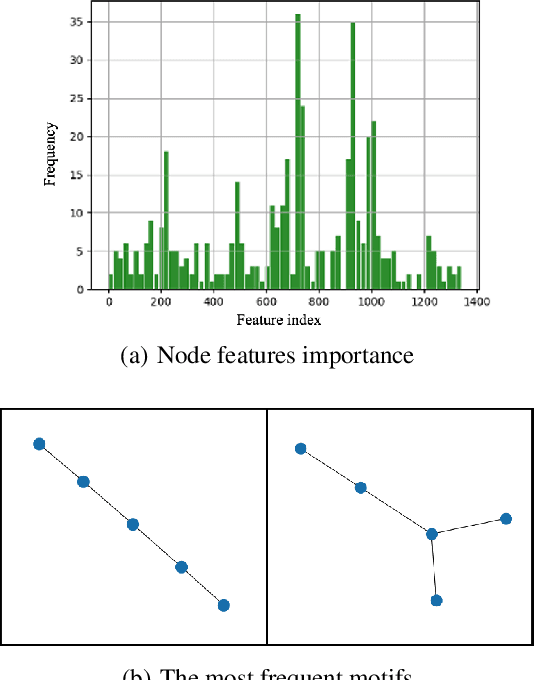
Double-strand DNA breaks (DSBs) are a form of DNA damage that can cause abnormal chromosomal rearrangements. Recent technologies based on high-throughput experiments have obvious high costs and technical challenges.Therefore, we design a graph neural network based method to predict DSBs (GraphDSB), using DNA sequence features and chromosome structure information. In order to improve the expression ability of the model, we introduce Jumping Knowledge architecture and several effective structural encoding methods. The contribution of structural information to the prediction of DSBs is verified by the experiments on datasets from normal human epidermal keratinocytes (NHEK) and chronic myeloid leukemia cell line (K562), and the ablation studies further demonstrate the effectiveness of the designed components in the proposed GraphDSB framework. Finally, we use GNNExplainer to analyze the contribution of node features and topology to DSBs prediction, and proved the high contribution of 5-mer DNA sequence features and two chromatin interaction modes.
Domain-Adversarial Multi-Task Framework for Novel Therapeutic Property Prediction of Compounds
Sep 28, 2018

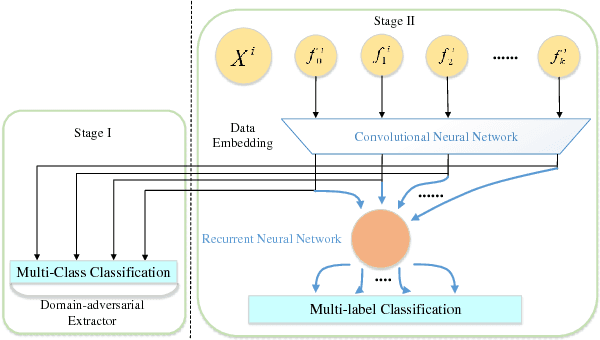
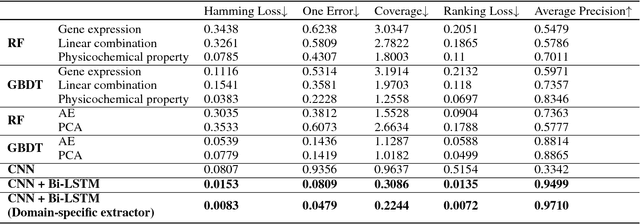
With the rapid development of high-throughput technologies, parallel acquisition of large-scale drug-informatics data provides huge opportunities to improve pharmaceutical research and development. One significant application is the purpose prediction of small molecule compounds, aiming to specify therapeutic properties of extensive purpose-unknown compounds and to repurpose novel therapeutic properties of FDA-approved drugs. Such problem is very challenging since compound attributes contain heterogeneous data with various feature patterns such as drug fingerprint, drug physicochemical property, drug perturbation gene expression. Moreover, there is complex nonlinear dependency among heterogeneous data. In this paper, we propose a novel domain-adversarial multi-task framework for integrating shared knowledge from multiple domains. The framework utilizes the adversarial strategy to effectively learn target representations and models their nonlinear dependency. Experiments on two real-world datasets illustrate that the performance of our approach obtains an obvious improvement over competitive baselines. The novel therapeutic properties of purpose-unknown compounds we predicted are mostly reported or brought to the clinics. Furthermore, our framework can integrate various attributes beyond the three domains examined here and can be applied in the industry for screening the purpose of huge amounts of as yet unidentified compounds. Source codes of this paper are available on Github.