 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDDA-Net: Accurate TDD Channel Estimation via Deep Unfolding the Doppler-Delay-Angle Representation of Channel Signals
Apr 07, 2026In TDD massive MIMO systems, channel estimation under sparse frequency-hopping pilots is challenging: each snapshot captures only one narrow pilot block that hops across frequency, with tens of milliseconds between adjacent snapshots. Finite-window leakage and off-grid effects weaken the ideal Doppler-delay-angle (DDA) sparsity, limiting both classical sparse recovery and purely data-driven approaches lacking an explicit structured transform-domain model. We propose DDA-Net, a model-driven 3D deep unfolding network for joint multi-snapshot channel state reconstruction. DDA-Net unfolds an ADMM formulation with an exact closed-form data-consistency update that avoids tensor inversion, learns the prior via a lightweight Doppler-domain denoiser, and uses delay oversampling to reduce basis mismatch. On QuaDRiGa UMa-NLOS, DDA-Net improves NMSE over the best baseline by more than 5 dB at 10 dB SNR, and retains a lead of about 1.5 dB under zero-shot testing on 3GPP CDL-B channels at the same SNR. Ablation studies show that window-level 3D processing is necessary across scenarios, while Doppler parameterization adds in-distribution gains and recovers a clear lead under scenario shift after few-shot fine-tuning with only 20 target-domain samples. These results demonstrate that combining exact physical data consistency with a learned DDA-domain prior is an effective and sample-efficient approach to channel state acquisition under sparse frequency-hopping pilots.
Covering-radius and Collinearity- Minimizing Pilots for Channel Estimation in TDD Systems
Apr 07, 2026This letter studies pilot design for orthogonal frequency-division multiplexing-based time-division duplex (TDD) systems under a sliding-window latest-slot recovery framework that jointly exploits delay--Doppler sparsity across recent slots. Under contiguous-subband and fairness constraints, this viewpoint naturally leads to a geometry-aware time--frequency joint pilot assignment. We show that effective patterns should balance grid coverage and redundant-collinearity suppression, with an additional symmetry-avoidance refinement when complete collinearity elimination is infeasible. Based on these principles, we formulate a mixed-integer construction method compatible with practical TDD allocation. Numerical results show that minimum-coverage-radius and collinearity-control (MCC) pattern improves both surrogate geometry metrics and latest-slot recovery performance.
Prior-Guided Symbolic Regression: Towards Scientific Consistency in Equation Discovery
Feb 16, 2026Symbolic Regression (SR) aims to discover interpretable equations from observational data, with the potential to reveal underlying principles behind natural phenomena. However, existing approaches often fall into the Pseudo-Equation Trap: producing equations that fit observations well but remain inconsistent with fundamental scientific principles. A key reason is that these approaches are dominated by empirical risk minimization, lacking explicit constraints to ensure scientific consistency. To bridge this gap, we propose PG-SR, a prior-guided SR framework built upon a three-stage pipeline consisting of warm-up, evolution, and refinement. Throughout the pipeline, PG-SR introduces a prior constraint checker that explicitly encodes domain priors as executable constraint programs, and employs a Prior Annealing Constrained Evaluation (PACE) mechanism during the evolution stage to progressively steer discovery toward scientifically consistent regions. Theoretically, we prove that PG-SR reduces the Rademacher complexity of the hypothesis space, yielding tighter generalization bounds and establishing a guarantee against pseudo-equations. Experimentally, PG-SR outperforms state-of-the-art baselines across diverse domains, maintaining robustness to varying prior quality, noisy data, and data scarcity.
WFR-MFM: One-Step Inference for Dynamic Unbalanced Optimal Transport
Jan 28, 2026Reconstructing dynamical evolution from limited observations is a fundamental challenge in single-cell biology, where dynamic unbalanced optimal transport provides a principled framework for modeling coupled transport and mass variation. However, existing approaches rely on trajectory simulation at inference time, making inference a key bottleneck for scalable applications. In this work, we propose a mean-flow framework for unbalanced flow matching that summarizes both transport and mass-growth dynamics over arbitrary time intervals using mean velocity and mass-growth fields, enabling fast one-step generation without trajectory simulation. To solve dynamic unbalanced optimal transport under the Wasserstein-Fisher-Rao geometry, we further build on this framework to develop Wasserstein-Fisher-Rao Mean Flow Matching (WFR-MFM). Across synthetic and real single-cell RNA sequencing datasets, WFR-MFM achieves orders-of-magnitude faster inference than a range of existing baselines while maintaining high predictive accuracy, and enables efficient perturbation response prediction on large synthetic datasets with thousands of conditions.
WFR-FM: Simulation-Free Dynamic Unbalanced Optimal Transport
Jan 11, 2026The Wasserstein-Fisher-Rao (WFR) metric extends dynamic optimal transport (OT) by coupling displacement with change of mass, providing a principled geometry for modeling unbalanced snapshot dynamics. Existing WFR solvers, however, are often unstable, computationally expensive, and difficult to scale. Here we introduce WFR Flow Matching (WFR-FM), a simulation-free training algorithm that unifies flow matching with dynamic unbalanced OT. Unlike classical flow matching which regresses only a transport vector field, WFR-FM simultaneously regresses a vector field for displacement and a scalar growth rate function for birth-death dynamics, yielding continuous flows under the WFR geometry. Theoretically, we show that minimizing the WFR-FM loss exactly recovers WFR geodesics. Empirically, WFR-FM yields more accurate and robust trajectory inference in single-cell biology, reconstructing consistent dynamics with proliferation and apoptosis, estimating time-varying growth fields, and applying to generative dynamics under imbalanced data. It outperforms state-of-the-art baselines in efficiency, stability, and reconstruction accuracy. Overall, WFR-FM establishes a unified and efficient paradigm for learning dynamical systems from unbalanced snapshots, where not only states but also mass evolve over time.
Variational Regularized Unbalanced Optimal Transport: Single Network, Least Action
May 17, 2025Recovering the dynamics from a few snapshots of a high-dimensional system is a challenging task in statistical physics and machine learning, with important applications in computational biology. Many algorithms have been developed to tackle this problem, based on frameworks such as optimal transport and the Schr\"odinger bridge. A notable recent framework is Regularized Unbalanced Optimal Transport (RUOT), which integrates both stochastic dynamics and unnormalized distributions. However, since many existing methods do not explicitly enforce optimality conditions, their solutions often struggle to satisfy the principle of least action and meet challenges to converge in a stable and reliable way. To address these issues, we propose Variational RUOT (Var-RUOT), a new framework to solve the RUOT problem. By incorporating the optimal necessary conditions for the RUOT problem into both the parameterization of the search space and the loss function design, Var-RUOT only needs to learn a scalar field to solve the RUOT problem and can search for solutions with lower action. We also examined the challenge of selecting a growth penalty function in the widely used Wasserstein-Fisher-Rao metric and proposed a solution that better aligns with biological priors in Var-RUOT. We validated the effectiveness of Var-RUOT on both simulated data and real single-cell datasets. Compared with existing algorithms, Var-RUOT can find solutions with lower action while exhibiting faster convergence and improved training stability.
Modeling Cell Dynamics and Interactions with Unbalanced Mean Field Schrödinger Bridge
May 16, 2025Modeling the dynamics from sparsely time-resolved snapshot data is crucial for understanding complex cellular processes and behavior. Existing methods leverage optimal transport, Schr\"odinger bridge theory, or their variants to simultaneously infer stochastic, unbalanced dynamics from snapshot data. However, these approaches remain limited in their ability to account for cell-cell interactions. This integration is essential in real-world scenarios since intercellular communications are fundamental life processes and can influence cell state-transition dynamics. To address this challenge, we formulate the Unbalanced Mean-Field Schr\"odinger Bridge (UMFSB) framework to model unbalanced stochastic interaction dynamics from snapshot data. Inspired by this framework, we further propose CytoBridge, a deep learning algorithm designed to approximate the UMFSB problem. By explicitly modeling cellular transitions, proliferation, and interactions through neural networks, CytoBridge offers the flexibility to learn these processes directly from data. The effectiveness of our method has been extensively validated using both synthetic gene regulatory data and real scRNA-seq datasets. Compared to existing methods, CytoBridge identifies growth, transition, and interaction patterns, eliminates false transitions, and reconstructs the developmental landscape with greater accuracy.
Improving the Euclidean Diffusion Generation of Manifold Data by Mitigating Score Function Singularity
May 15, 2025Euclidean diffusion models have achieved remarkable success in generative modeling across diverse domains, and they have been extended to manifold case in recent advances. Instead of explicitly utilizing the structure of special manifolds as studied in previous works, we investigate direct sampling of the Euclidean diffusion models for general manifold-constrained data in this paper. We reveal the multiscale singularity of the score function in the embedded space of manifold, which hinders the accuracy of diffusion-generated samples. We then present an elaborate theoretical analysis of the singularity structure of the score function by separating it along the tangential and normal directions of the manifold. To mitigate the singularity and improve the sampling accuracy, we propose two novel methods: (1) Niso-DM, which introduces non-isotropic noise along the normal direction to reduce scale discrepancies, and (2) Tango-DM, which trains only the tangential component of the score function using a tangential-only loss function. Numerical experiments demonstrate that our methods achieve superior performance on distributions over various manifolds with complex geometries.
Riemannian Denoising Diffusion Probabilistic Models
May 07, 2025We propose Riemannian Denoising Diffusion Probabilistic Models (RDDPMs) for learning distributions on submanifolds of Euclidean space that are level sets of functions, including most of the manifolds relevant to applications. Existing methods for generative modeling on manifolds rely on substantial geometric information such as geodesic curves or eigenfunctions of the Laplace-Beltrami operator and, as a result, they are limited to manifolds where such information is available. In contrast, our method, built on a projection scheme, can be applied to more general manifolds, as it only requires being able to evaluate the value and the first order derivatives of the function that defines the submanifold. We provide a theoretical analysis of our method in the continuous-time limit, which elucidates the connection between our RDDPMs and score-based generative models on manifolds. The capability of our method is demonstrated on datasets from previous studies and on new datasets sampled from two high-dimensional manifolds, i.e. $\mathrm{SO}(10)$ and the configuration space of molecular system alanine dipeptide with fixed dihedral angle.
Integrating Dynamical Systems Modeling with Spatiotemporal scRNA-seq Data Analysis
Mar 14, 2025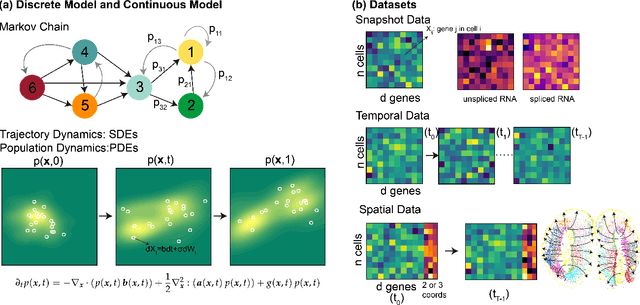
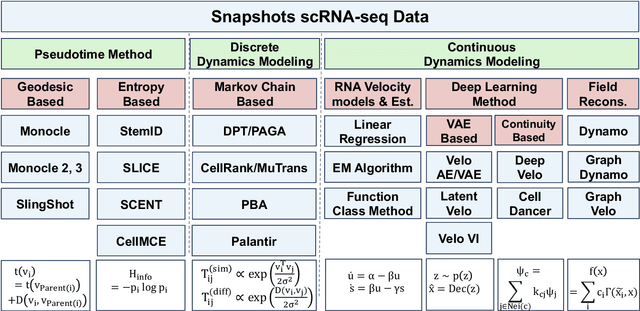
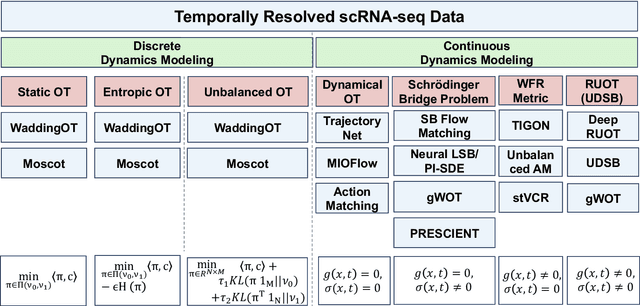

Understanding the dynamic nature of biological systems is fundamental to deciphering cellular behavior, developmental processes, and disease progression. Single-cell RNA sequencing (scRNA-seq) has provided static snapshots of gene expression, offering valuable insights into cellular states at a single time point. Recent advancements in temporally resolved scRNA-seq, spatial transcriptomics (ST), and time-series spatial transcriptomics (temporal-ST) have further revolutionized our ability to study the spatiotemporal dynamics of individual cells. These technologies, when combined with computational frameworks such as Markov chains, stochastic differential equations (SDEs), and generative models like optimal transport and Schr\"odinger bridges, enable the reconstruction of dynamic cellular trajectories and cell fate decisions. This review discusses how these dynamical system approaches offer new opportunities to model and infer cellular dynamics from a systematic perspective.