 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCross-Modality Controlled Molecule Generation with Diffusion Language Model
Aug 20, 2025



Current SMILES-based diffusion models for molecule generation typically support only unimodal constraint. They inject conditioning signals at the start of the training process and require retraining a new model from scratch whenever the constraint changes. However, real-world applications often involve multiple constraints across different modalities, and additional constraints may emerge over the course of a study. This raises a challenge: how to extend a pre-trained diffusion model not only to support cross-modality constraints but also to incorporate new ones without retraining. To tackle this problem, we propose the Cross-Modality Controlled Molecule Generation with Diffusion Language Model (CMCM-DLM), demonstrated by two distinct cross modalities: molecular structure and chemical properties. Our approach builds upon a pre-trained diffusion model, incorporating two trainable modules, the Structure Control Module (SCM) and the Property Control Module (PCM), and operates in two distinct phases during the generation process. In Phase I, we employs the SCM to inject structural constraints during the early diffusion steps, effectively anchoring the molecular backbone. Phase II builds on this by further introducing PCM to guide the later stages of inference to refine the generated molecules, ensuring their chemical properties match the specified targets. Experimental results on multiple datasets demonstrate the efficiency and adaptability of our approach, highlighting CMCM-DLM's significant advancement in molecular generation for drug discovery applications.
ToolFactory: Automating Tool Generation by Leveraging LLM to Understand REST API Documentations
Jan 28, 2025LLM-based tool agents offer natural language interfaces, enabling users to seamlessly interact with computing services. While REST APIs are valuable resources for building such agents, they must first be transformed into AI-compatible tools. Automatically generating AI-compatible tools from REST API documents can greatly streamline tool agent development and minimize user learning curves. However, API documentation often suffers from a lack of standardization, inconsistent schemas, and incomplete information. To address these issues, we developed \textbf{ToolFactory}, an open-source pipeline for automating tool generation from unstructured API documents. To enhance the reliability of the developed tools, we implemented an evaluation method to diagnose errors. Furthermore, we built a knowledge base of verified tools, which we leveraged to infer missing information from poorly documented APIs. We developed the API Extraction Benchmark, comprising 167 API documents and 744 endpoints in various formats, and designed a JSON schema to annotate them. This annotated dataset was utilized to train and validate ToolFactory. The experimental results highlight the effectiveness of ToolFactory. We also demonstrated ToolFactory by creating a domain-specific AI agent for glycomaterials research. ToolFactory exhibits significant potential for facilitating the seamless integration of scientific REST APIs into AI workflows.
Multiple Abstraction Level Retrieve Augment Generation
Jan 28, 2025



A Retrieval-Augmented Generation (RAG) model powered by a large language model (LLM) provides a faster and more cost-effective solution for adapting to new data and knowledge. It also delivers more specialized responses compared to pre-trained LLMs. However, most existing approaches rely on retrieving prefix-sized chunks as references to support question-answering (Q/A). This approach is often deployed to address information needs at a single level of abstraction, as it struggles to generate answers across multiple levels of abstraction. In an RAG setting, while LLMs can summarize and answer questions effectively when provided with sufficient details, retrieving excessive information often leads to the 'lost in the middle' problem and exceeds token limitations. We propose a novel RAG approach that uses chunks of multiple abstraction levels (MAL), including multi-sentence-level, paragraph-level, section-level, and document-level. The effectiveness of our approach is demonstrated in an under-explored scientific domain of Glycoscience. Compared to traditional single-level RAG approaches, our approach improves AI evaluated answer correctness of Q/A by 25.739\% on Glyco-related papers.
Reflections from the 2024 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry
Nov 20, 2024
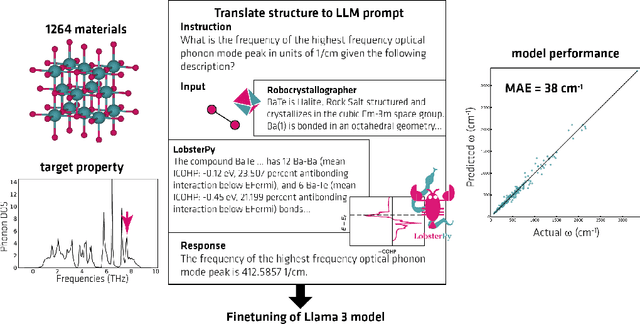
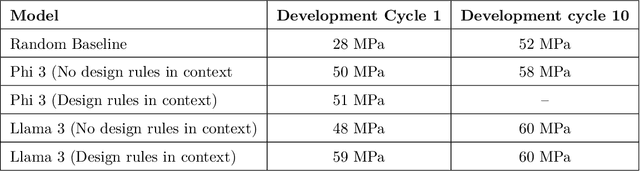
Here, we present the outcomes from the second Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry, which engaged participants across global hybrid locations, resulting in 34 team submissions. The submissions spanned seven key application areas and demonstrated the diverse utility of LLMs for applications in (1) molecular and material property prediction; (2) molecular and material design; (3) automation and novel interfaces; (4) scientific communication and education; (5) research data management and automation; (6) hypothesis generation and evaluation; and (7) knowledge extraction and reasoning from scientific literature. Each team submission is presented in a summary table with links to the code and as brief papers in the appendix. Beyond team results, we discuss the hackathon event and its hybrid format, which included physical hubs in Toronto, Montreal, San Francisco, Berlin, Lausanne, and Tokyo, alongside a global online hub to enable local and virtual collaboration. Overall, the event highlighted significant improvements in LLM capabilities since the previous year's hackathon, suggesting continued expansion of LLMs for applications in materials science and chemistry research. These outcomes demonstrate the dual utility of LLMs as both multipurpose models for diverse machine learning tasks and platforms for rapid prototyping custom applications in scientific research.
Theoretical Corrections and the Leveraging of Reinforcement Learning to Enhance Triangle Attack
Nov 18, 2024



Adversarial examples represent a serious issue for the application of machine learning models in many sensitive domains. For generating adversarial examples, decision based black-box attacks are one of the most practical techniques as they only require query access to the model. One of the most recently proposed state-of-the-art decision based black-box attacks is Triangle Attack (TA). In this paper, we offer a high-level description of TA and explain potential theoretical limitations. We then propose a new decision based black-box attack, Triangle Attack with Reinforcement Learning (TARL). Our new attack addresses the limits of TA by leveraging reinforcement learning. This creates an attack that can achieve similar, if not better, attack accuracy than TA with half as many queries on state-of-the-art classifiers and defenses across ImageNet and CIFAR-10.
Uncertainty Quantification for Clinical Outcome Predictions with (Large) Language Models
Nov 05, 2024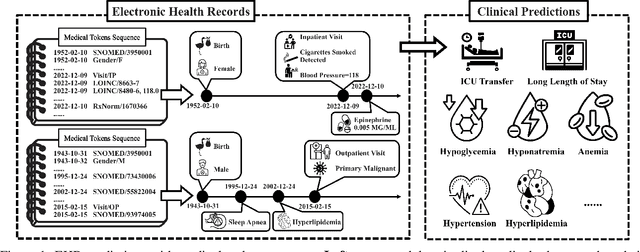



To facilitate healthcare delivery, language models (LMs) have significant potential for clinical prediction tasks using electronic health records (EHRs). However, in these high-stakes applications, unreliable decisions can result in high costs due to compromised patient safety and ethical concerns, thus increasing the need for good uncertainty modeling of automated clinical predictions. To address this, we consider the uncertainty quantification of LMs for EHR tasks in white- and black-box settings. We first quantify uncertainty in white-box models, where we can access model parameters and output logits. We show that an effective reduction of model uncertainty can be achieved by using the proposed multi-tasking and ensemble methods in EHRs. Continuing with this idea, we extend our approach to black-box settings, including popular proprietary LMs such as GPT-4. We validate our framework using longitudinal clinical data from more than 6,000 patients in ten clinical prediction tasks. Results show that ensembling methods and multi-task prediction prompts reduce uncertainty across different scenarios. These findings increase the transparency of the model in white-box and black-box settings, thus advancing reliable AI healthcare.
Question Rephrasing for Quantifying Uncertainty in Large Language Models: Applications in Molecular Chemistry Tasks
Aug 07, 2024Uncertainty quantification enables users to assess the reliability of responses generated by large language models (LLMs). We present a novel Question Rephrasing technique to evaluate the input uncertainty of LLMs, which refers to the uncertainty arising from equivalent variations of the inputs provided to LLMs. This technique is integrated with sampling methods that measure the output uncertainty of LLMs, thereby offering a more comprehensive uncertainty assessment. We validated our approach on property prediction and reaction prediction for molecular chemistry tasks.
Deep-learning Optical Flow Outperforms PIV in Obtaining Velocity Fields from Active Nematics
Apr 26, 2024



Deep learning-based optical flow (DLOF) extracts features in adjacent video frames with deep convolutional neural networks. It uses those features to estimate the inter-frame motions of objects at the pixel level. In this article, we evaluate the ability of optical flow to quantify the spontaneous flows of MT-based active nematics under different labeling conditions. We compare DLOF against the commonly used technique, particle imaging velocimetry (PIV). We obtain flow velocity ground truths either by performing semi-automated particle tracking on samples with sparsely labeled filaments, or from passive tracer beads. We find that DLOF produces significantly more accurate velocity fields than PIV for densely labeled samples. We show that the breakdown of PIV arises because the algorithm cannot reliably distinguish contrast variations at high densities, particularly in directions parallel to the nematic director. DLOF overcomes this limitation. For sparsely labeled samples, DLOF and PIV produce results with similar accuracy, but DLOF gives higher-resolution fields. Our work establishes DLOF as a versatile tool for measuring fluid flows in a broad class of active, soft, and biophysical systems.
Solvent-Aware 2D NMR Prediction: Leveraging Multi-Tasking Training and Iterative Self-Training Strategies
Mar 17, 2024



Nuclear magnetic resonance (NMR) spectroscopy plays a pivotal role in various scientific fields, offering insights into structural information, electronic properties and dynamic behaviors of molecules. Accurate NMR spectrum prediction efficiently produces candidate molecules, enabling chemists to compare them with actual experimental spectra. This process aids in confirming molecular structures or pinpointing discrepancies, guiding further investigation. Machine Learning (ML) has then emerged as a promising alternative approach for predicting atomic NMR chemical shits of molecules given their structures. Although significant progresses have been made in predicting one-dimensional (1D) NMR, two-dimensional (2D) NMR prediction via ML remains a challenge due to the lack of annotated NMR training datasets. To address this gap, we propose an iterative self-training (IST) approach to train a deep learning model for predicting atomic 2DNMR shifts and assigning peaks in experimental spectra. Our model undergoes an initial pre-training phase employing a Multi-Task Training (MTT) approach, which simultaneously leverages annotated 1D NMR datasets of both $^{1}\text{H}$ and $^{13}\text{C}$ spectra to enhance its understanding of NMR spectra. Subsequently, the pre-trained model is utilized to generate pseudo-annotations for unlabelled 2D NMR spectra, which are subsequently used to refine the 2D NMR prediction model. Our approach iterates between annotated unlabelled 2D NMR data and refining our 2D NMR prediction model until convergence. Finally, our model is able to not only accurately predict 2D NMR but also annotate peaks in experimental 2D NMR spectra. Experimental results show that our model is capable of accurately handling medium-sized and large molecules, including polysaccharides, underscoring its effectiveness.
Graph Multi-Similarity Learning for Molecular Property Prediction
Feb 02, 2024



Enhancing accurate molecular property prediction relies on effective and proficient representation learning. It is crucial to incorporate diverse molecular relationships characterized by multi-similarity (self-similarity and relative similarities) between molecules. However, current molecular representation learning methods fall short in exploring multi-similarity and often underestimate the complexity of relationships between molecules. Additionally, previous multi-similarity approaches require the specification of positive and negative pairs to attribute distinct predefined weights to different relative similarities, which can introduce potential bias. In this work, we introduce Graph Multi-Similarity Learning for Molecular Property Prediction (GraphMSL) framework, along with a novel approach to formulate a generalized multi-similarity metric without the need to define positive and negative pairs. In each of the chemical modality spaces (e.g.,molecular depiction image, fingerprint, NMR, and SMILES) under consideration, we first define a self-similarity metric (i.e., similarity between an anchor molecule and another molecule), and then transform it into a generalized multi-similarity metric for the anchor through a pair weighting function. GraphMSL validates the efficacy of the multi-similarity metric across MoleculeNet datasets. Furthermore, these metrics of all modalities are integrated into a multimodal multi-similarity metric, which showcases the potential to improve the performance. Moreover, the focus of the model can be redirected or customized by altering the fusion function. Last but not least, GraphMSL proves effective in drug discovery evaluations through post-hoc analyses of the learnt representations.