 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTensor Decomposition Networks for Fast Machine Learning Interatomic Potential Computations
Jul 01, 2025$\rm{SO}(3)$-equivariant networks are the dominant models for machine learning interatomic potentials (MLIPs). The key operation of such networks is the Clebsch-Gordan (CG) tensor product, which is computationally expensive. To accelerate the computation, we develop tensor decomposition networks (TDNs) as a class of approximately equivariant networks whose CG tensor products are replaced by low-rank tensor decompositions, such as the CANDECOMP/PARAFAC (CP) decomposition. With the CP decomposition, we prove (i) a uniform bound on the induced error of $\rm{SO}(3)$-equivariance, and (ii) the universality of approximating any equivariant bilinear map. To further reduce the number of parameters, we propose path-weight sharing that ties all multiplicity-space weights across the $O(L^3)$ CG paths into a single path without compromising equivariance, where $L$ is the maximum angular degree. The resulting layer acts as a plug-and-play replacement for tensor products in existing networks, and the computational complexity of tensor products is reduced from $O(L^6)$ to $O(L^4)$. We evaluate TDNs on PubChemQCR, a newly curated molecular relaxation dataset containing 105 million DFT-calculated snapshots. We also use existing datasets, including OC20, and OC22. Results show that TDNs achieve competitive performance with dramatic speedup in computations.
PubChemQC B3LYP/6-31G*//PM6 dataset: the Electronic Structures of 86 Million Molecules using B3LYP/6-31G* calculations
May 29, 2023This article presents the "PubChemQC B3LYP/6-31G*//PM6" dataset, containing electronic properties of 85,938,443 molecules. It includes orbitals, orbital energies, total energies, dipole moments, and other relevant properties. The dataset encompasses a wide range of molecules, from essential compounds to biomolecules up to 1000 molecular weight, covering 94.0% of the original PubChem Compound catalog (as of August 29, 2016). The electronic properties were calculated using the B3LYP/6-31G* and PM6 methods. The dataset is available in three formats: (i) GAMESS quantum chemistry program files, (ii) selected JSON output files, and (iii) a PostgreSQL database, enabling researchers to query molecular properties. Five sub-datasets offer more specific data. The first two subsets include molecules with C, H, O, and N, under 300 and 500 molecular weight respectively. The third and fourth subsets contain C, H, N, O, P, S, F, and Cl, under 300 and 500 molecular weight respectively. The fifth subset includes C, H, N, O, P, S, F, Cl, Na, K, Mg, and Ca, under 500 molecular weight. Coefficients of determination ranged from 0.892 (CHON500) to 0.803 (whole) for the HOMO-LUMO energy gap. These findings represent extensive investigations and can be utilized for drug discovery, material science, and other applications. The datasets are available under the Creative Commons Attribution 4.0 International license at https://nakatamaho.riken.jp/pubchemqc.riken.jp/b3lyp_pm6_datasets.html.
Molecule3D: A Benchmark for Predicting 3D Geometries from Molecular Graphs
Sep 30, 2021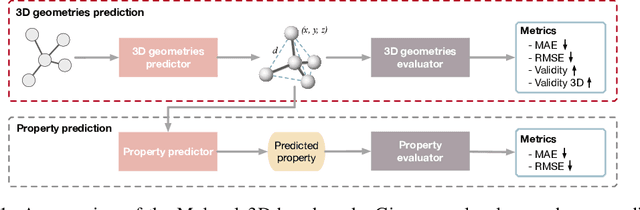
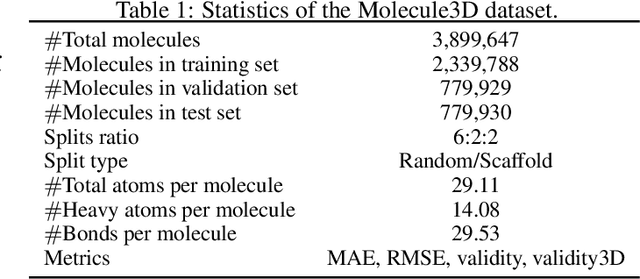
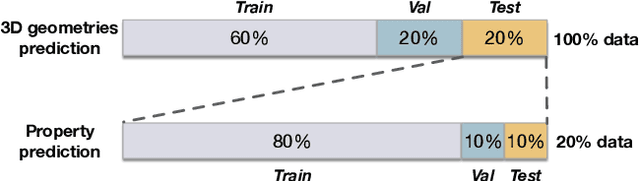
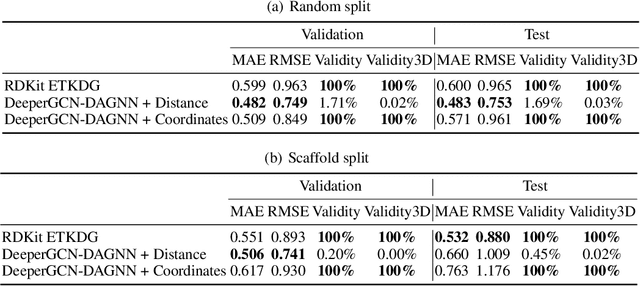
Graph neural networks are emerging as promising methods for modeling molecular graphs, in which nodes and edges correspond to atoms and chemical bonds, respectively. Recent studies show that when 3D molecular geometries, such as bond lengths and angles, are available, molecular property prediction tasks can be made more accurate. However, computing of 3D molecular geometries requires quantum calculations that are computationally prohibitive. For example, accurate calculation of 3D geometries of a small molecule requires hours of computing time using density functional theory (DFT). Here, we propose to predict the ground-state 3D geometries from molecular graphs using machine learning methods. To make this feasible, we develop a benchmark, known as Molecule3D, that includes a dataset with precise ground-state geometries of approximately 4 million molecules derived from DFT. We also provide a set of software tools for data processing, splitting, training, and evaluation, etc. Specifically, we propose to assess the error and validity of predicted geometries using four metrics. We implement two baseline methods that either predict the pairwise distance between atoms or atom coordinates in 3D space. Experimental results show that, compared with generating 3D geometries with RDKit, our method can achieve comparable prediction accuracy but with much smaller computational costs. Our Molecule3D is available as a module of the MoleculeX software library (https://github.com/divelab/MoleculeX).
OGB-LSC: A Large-Scale Challenge for Machine Learning on Graphs
Mar 17, 2021
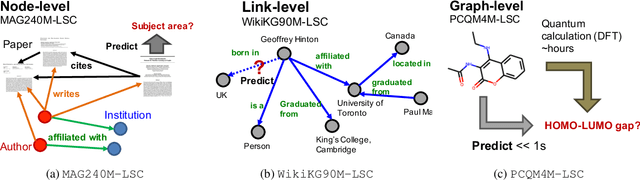
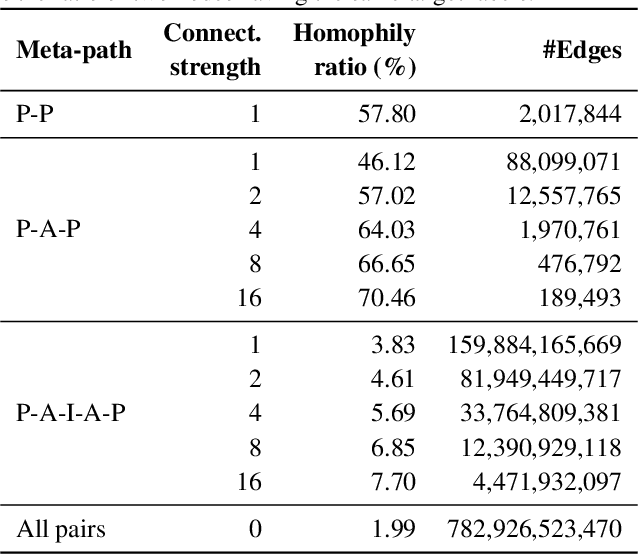
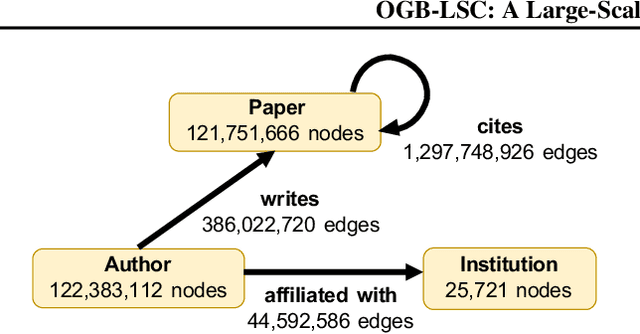
Enabling effective and efficient machine learning (ML) over large-scale graph data (e.g., graphs with billions of edges) can have a huge impact on both industrial and scientific applications. However, community efforts to advance large-scale graph ML have been severely limited by the lack of a suitable public benchmark. For KDD Cup 2021, we present OGB Large-Scale Challenge (OGB-LSC), a collection of three real-world datasets for advancing the state-of-the-art in large-scale graph ML. OGB-LSC provides graph datasets that are orders of magnitude larger than existing ones and covers three core graph learning tasks -- link prediction, graph regression, and node classification. Furthermore, OGB-LSC provides dedicated baseline experiments, scaling up expressive graph ML models to the massive datasets. We show that the expressive models significantly outperform simple scalable baselines, indicating an opportunity for dedicated efforts to further improve graph ML at scale. Our datasets and baseline code are released and maintained as part of our OGB initiative (Hu et al., 2020). We hope OGB-LSC at KDD Cup 2021 can empower the community to discover innovative solutions for large-scale graph ML.