 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePeriodic Topological Deep Learning for Polymer Design and Discovery
May 26, 2026Polymers underpin applications across energy, healthcare, and materials science, yet their vast chemical space makes systematic discovery challenging. Most machine learning approaches represent polymers as molecular graphs of a single repeating unit, thereby missing both the periodicity of polymer chains and many-body interactions beyond pairwise bonds. We introduce Periodic-TDL, a deep learning framework built on periodic Vietoris-Rips complexes that capture many-body interactions across multiple spatial scales, followed by a hierarchical simplicial message-passing (HSMP) encoder that propagates information from long-range interactions to covalent bonds, yielding representations enriched by higher-order topological features. Periodic-TDL outperforms all state-of-the-art models across polymer property prediction tasks spanning electronic, optical, physical, and thermal targets. Furthermore, we quantitatively validate how ester-to-amide substitution and $α$-methylation enhance thermal stability. Using a computationally synthesized dataset of 48,208 structures-generated via systematic substitution of acrylate and acrylamide polymers-we observed a mean $T_g$ increase of $\sim 55^\circ$C for ester-to-amide substitutions and $\sim 14^\circ$C for backbone $α$-methylation across matched polymer pairs. To verify these predicted trends, we use our Periodic-TDL model to analyze six novel polymer pairs from independent experimental measurements, including three newly synthesized polymers previously unreported in the literature. The experimental data successfully confirmed the model's predictions. Ultimately, these findings demonstrate that Periodic-TDL captures the underlying physical effects of specific functional group modifications, rather than merely optimizing predictive performance on benchmark datasets.
Full-Spectrum Graph Neural Network: Expressive and Scalable
May 07, 2026It is well established that spectral graph neural networks (GNNs) can universally approximate node signals; however, their expressive power remains bounded by the 1-dimensional Weisfeiler-Lehman test, which is mirrored in their lack of universality for higher-order signals. To go beyond this bound, we propose the Full-Spectrum GNN (FSpecGNN), a second-order generalization of classical spectral GNNs. FSpecGNN advances spectral filtering in two perspectives: (1) it lifts the signal from the node domain to the node-pair domain; and (2) it extends the univariate spectral filter over eigenvalues to a bivariate filter over eigenvalue pairs. We show that classical spectral GNNs arise as a diagonal special case of FSpecGNN, and prove that FSpecGNN can be at most as expressive as Local 2-GNN while universally approximating node-pair signals, the latter being particularly beneficial for heterophilic graph learning. Moreover, FSpecGNN admits scalable implementations that avoid explicit node-pair-level computations; combined with a low-rank approximation that reduces full-spectrum convolution to a combination of polynomial spectral filters, it enables learning on large graphs. Empirically, FSpecGNN validates the predicted expressivity and delivers strong performance on heterophilic benchmarks.
Sheaf Neural Networks on SPD Manifolds: Second-Order Geometric Representation Learning
Apr 22, 2026Graph neural networks face two fundamental challenges rooted in the linear structure of Euclidean vector spaces: (1) Current architectures represent geometry through vectors (directions, gradients), yet many tasks require matrix-valued representations that capture relationships between directions-such as how atomic orientations covary in a molecule. These second-order representations are naturally captured by points on the symmetric positive definite matrices (SPD) manifold; (2) Standard message passing applies shared transformations across edges. Sheaf neural networks address this via edge-specific transformations, but existing formulations remain confined to vector spaces and therefore cannot propagate matrix-valued features. We address both challenges by developing the first sheaf neural network operates natively on the SPD manifold. Our key insight is that the SPD manifold admits a Lie group structure, enabling well-posed analogs of sheaf operators without projecting to Euclidean space. Theoretically, we prove that SPD-valued sheaves are strictly more expressive than Euclidean sheaves: they admit consistent configurations (global sections) that vector-valued sheaves cannot represent, directly translating to richer learned representations. Empirically, our sheaf convolution transforms effectively rank-1 directional inputs into full-rank matrices encoding local geometric structure. Our dual-stream architecture achieves SOTA on 6/7 MoleculeNet benchmarks, with the sheaf framework providing consistent depth robustness.
Topology-Aware Multiscale Mixture of Experts for Efficient Molecular Property Prediction
Jan 19, 2026Many molecular properties depend on 3D geometry, where non-covalent interactions, stereochemical effects, and medium- to long-range forces are determined by spatial distances and angles that cannot be uniquely captured by a 2D bond graph. Yet most 3D molecular graph neural networks still rely on globally fixed neighborhood heuristics, typically defined by distance cutoffs and maximum neighbor limits, to define local message-passing neighborhoods, leading to rigid, data-agnostic interaction budgets. We propose Multiscale Interaction Mixture of Experts (MI-MoE) to adapt interaction modeling across geometric regimes. Our contributions are threefold: (1) we introduce a distance-cutoff expert ensemble that explicitly captures short-, mid-, and long-range interactions without committing to a single cutoff; (2) we design a topological gating encoder that routes inputs to experts using filtration-based descriptors, including persistent homology features, summarizing how connectivity evolves across radii; and (3) we show that MI-MoE is a plug-in module that consistently improves multiple strong 3D molecular backbones across diverse molecular and polymer property prediction benchmark datasets, covering both regression and classification tasks. These results highlight topology-aware multiscale routing as an effective principle for 3D molecular graph learning.
A roadmap for curvature-based geometric data analysis and learning
Oct 26, 2025Geometric data analysis and learning has emerged as a distinct and rapidly developing research area, increasingly recognized for its effectiveness across diverse applications. At the heart of this field lies curvature, a powerful and interpretable concept that captures intrinsic geometric structure and underpins numerous tasks, from community detection to geometric deep learning. A wide range of discrete curvature models have been proposed for various data representations, including graphs, simplicial complexes, cubical complexes, and point clouds sampled from manifolds. These models not only provide efficient characterizations of data geometry but also constitute essential components in geometric learning frameworks. In this paper, we present the first comprehensive review of existing discrete curvature models, covering their mathematical foundations, computational formulations, and practical applications in data analysis and learning. In particular, we discuss discrete curvature from both Riemannian and metric geometry perspectives and propose a systematic pipeline for curvature-driven data analysis. We further examine the corresponding computational algorithms across different data representations, offering detailed comparisons and insights. Finally, we review state-of-the-art applications of curvature in both supervised and unsupervised learning. This survey provides a conceptual and practical roadmap for researchers to gain a better understanding of discrete curvature as a fundamental tool for geometric understanding and learning.
Quotient Complex Transformer (QCformer) for Perovskite Data Analysis
May 14, 2025The discovery of novel functional materials is crucial in addressing the challenges of sustainable energy generation and climate change. Hybrid organic-inorganic perovskites (HOIPs) have gained attention for their exceptional optoelectronic properties in photovoltaics. Recently, geometric deep learning, particularly graph neural networks (GNNs), has shown strong potential in predicting material properties and guiding material design. However, traditional GNNs often struggle to capture the periodic structures and higher-order interactions prevalent in such systems. To address these limitations, we propose a novel representation based on quotient complexes (QCs) and introduce the Quotient Complex Transformer (QCformer) for material property prediction. A material structure is modeled as a quotient complex, which encodes both pairwise and many-body interactions via simplices of varying dimensions and captures material periodicity through a quotient operation. Our model leverages higher-order features defined on simplices and processes them using a simplex-based Transformer module. We pretrain QCformer on benchmark datasets such as the Materials Project and JARVIS, and fine-tune it on HOIP datasets. The results show that QCformer outperforms state-of-the-art models in HOIP property prediction, demonstrating its effectiveness. The quotient complex representation and QCformer model together contribute a powerful new tool for predictive modeling of perovskite materials.
Rhomboid Tiling for Geometric Graph Deep Learning
May 14, 2025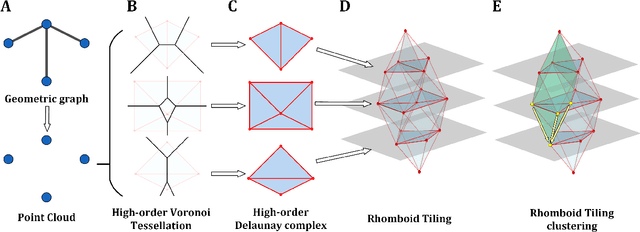
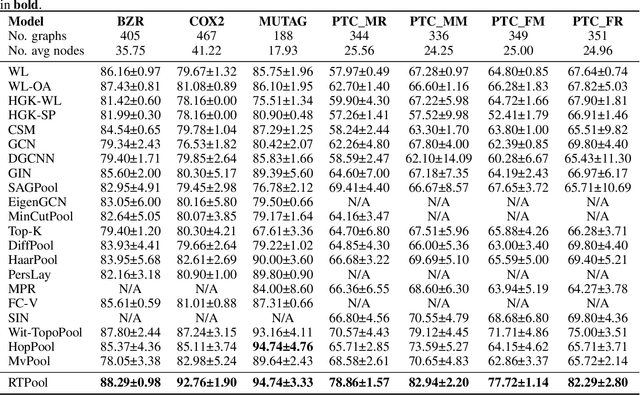
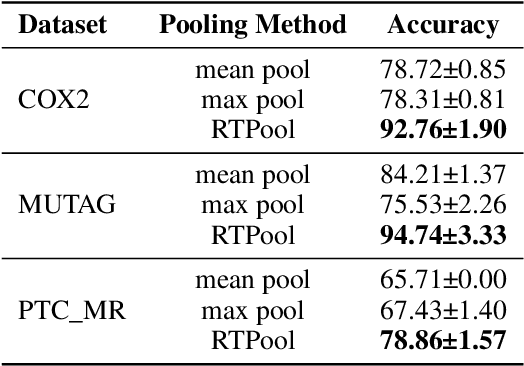
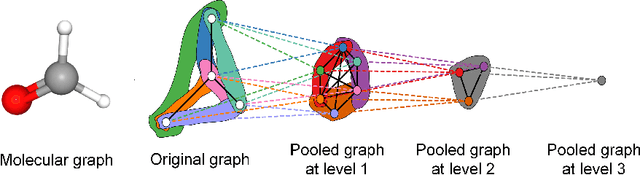
Graph Neural Networks (GNNs) have proven effective for learning from graph-structured data through their neighborhood-based message passing framework. Many hierarchical graph clustering pooling methods modify this framework by introducing clustering-based strategies, enabling the construction of more expressive and powerful models. However, all of these message passing framework heavily rely on the connectivity structure of graphs, limiting their ability to capture the rich geometric features inherent in geometric graphs. To address this, we propose Rhomboid Tiling (RT) clustering, a novel clustering method based on the rhomboid tiling structure, which performs clustering by leveraging the complex geometric information of the data and effectively extracts its higher-order geometric structures. Moreover, we design RTPool, a hierarchical graph clustering pooling model based on RT clustering for graph classification tasks. The proposed model demonstrates superior performance, outperforming 21 state-of-the-art competitors on all the 7 benchmark datasets.
A cohomology-based Gromov-Hausdorff metric approach for quantifying molecular similarity
Nov 21, 2024We introduce, for the first time, a cohomology-based Gromov-Hausdorff ultrametric method to analyze 1-dimensional and higher-dimensional (co)homology groups, focusing on loops, voids, and higher-dimensional cavity structures in simplicial complexes, to address typical clustering questions arising in molecular data analysis. The Gromov-Hausdorff distance quantifies the dissimilarity between two metric spaces. In this framework, molecules are represented as simplicial complexes, and their cohomology vector spaces are computed to capture intrinsic topological invariants encoding loop and cavity structures. These vector spaces are equipped with a suitable distance measure, enabling the computation of the Gromov-Hausdorff ultrametric to evaluate structural dissimilarities. We demonstrate the methodology using organic-inorganic halide perovskite (OIHP) structures. The results highlight the effectiveness of this approach in clustering various molecular structures. By incorporating geometric information, our method provides deeper insights compared to traditional persistent homology techniques.
KA-GNN: Kolmogorov-Arnold Graph Neural Networks for Molecular Property Prediction
Oct 15, 2024Molecular property prediction is a crucial task in the process of Artificial Intelligence-Driven Drug Discovery (AIDD). The challenge of developing models that surpass traditional non-neural network methods continues to be a vibrant area of research. This paper presents a novel graph neural network model-the Kolmogorov-Arnold Network (KAN)-based Graph Neural Network (KA-GNN), which incorporates Fourier series, specifically designed for molecular property prediction. This model maintains the high interpretability characteristic of KAN methods while being extremely efficient in computational resource usage, making it an ideal choice for deployment in resource-constrained environments. Tested and validated on seven public datasets, KA-GNN has shown significant improvements in property predictions over the existing state-of-the-art (SOTA) benchmarks.
Molecular topological deep learning for polymer property prediction
Oct 07, 2024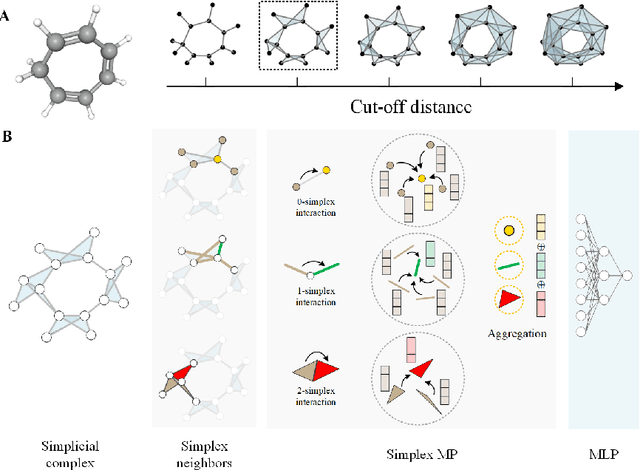
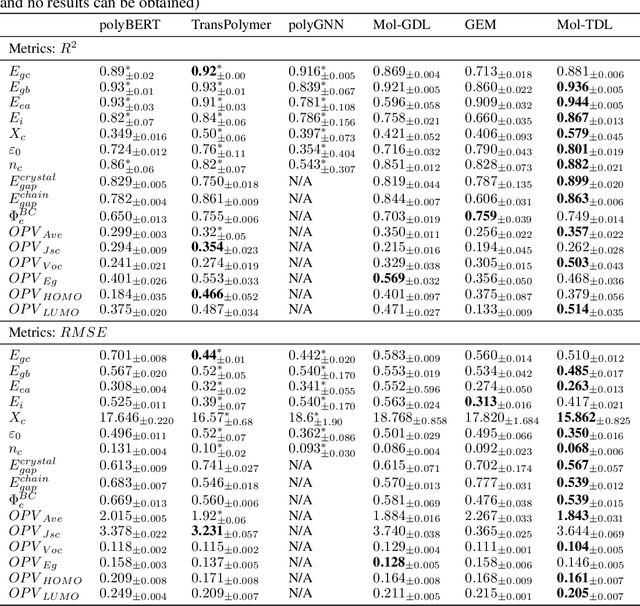
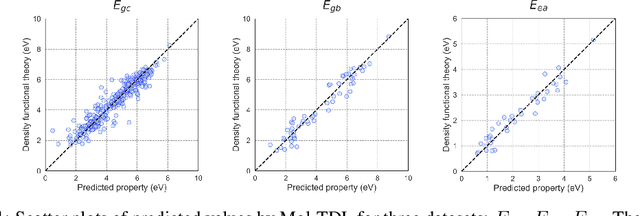
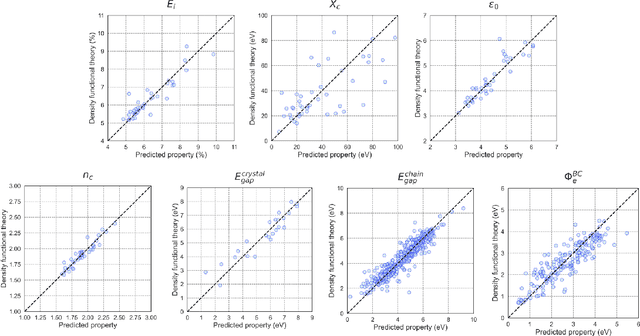
Accurate and efficient prediction of polymer properties is of key importance for polymer design. Traditional experimental tools and density function theory (DFT)-based simulations for polymer property evaluation, are both expensive and time-consuming. Recently, a gigantic amount of graph-based molecular models have emerged and demonstrated huge potential in molecular data analysis. Even with the great progresses, these models tend to ignore the high-order and mutliscale information within the data. In this paper, we develop molecular topological deep learning (Mol-TDL) for polymer property analysis. Our Mol-TDL incorporates both high-order interactions and multiscale properties into topological deep learning architecture. The key idea is to represent polymer molecules as a series of simplicial complices at different scales and build up simplical neural networks accordingly. The aggregated information from different scales provides a more accurate prediction of polymer molecular properties.