 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePersistent Sheaf Laplacian Analysis of Protein Stability and Solubility Changes upon Mutation
Jan 18, 2026Genetic mutations frequently disrupt protein structure, stability, and solubility, acting as primary drivers for a wide spectrum of diseases. Despite the critical importance of these molecular alterations, existing computational models often lack interpretability, and fail to integrate essential physicochemical interaction. To overcome these limitations, we propose SheafLapNet, a unified predictive framework grounded in the mathematical theory of Topological Deep Learning (TDL) and Persistent Sheaf Laplacian (PSL). Unlike standard Topological Data Analysis (TDA) tools such as persistent homology, which are often insensitive to heterogeneous information, PSL explicitly encodes specific physical and chemical information such as partial charges directly into the topological analysis. SheafLapNet synergizes these sheaf-theoretic invariants with advanced protein transformer features and auxiliary physical descriptors to capture intrinsic molecular interactions in a multiscale and mechanistic manner. To validate our framework, we employ rigorous benchmarks for both regression and classification tasks. For stability prediction, we utilize the comprehensive S2648 and S350 datasets. For solubility prediction, we employ the PON-Sol2 dataset, which provides annotations for increased, decreased, or neutral solubility changes. By integrating these multi-perspective features, SheafLapNet achieves state-of-the-art performance across these diverse benchmarks, demonstrating that sheaf-theoretic modeling significantly enhances both interpretability and generalizability in predicting mutation-induced structural and functional changes.
Curvature-enhanced Graph Convolutional Network for Biomolecular Interaction Prediction
Jun 23, 2023
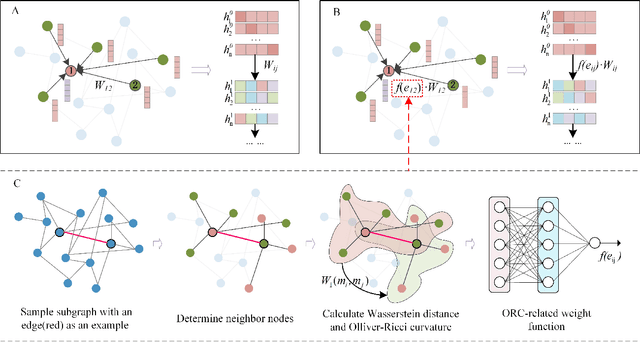
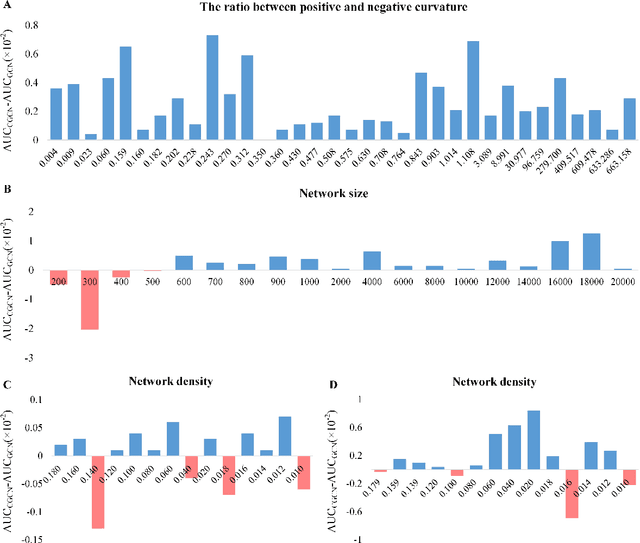
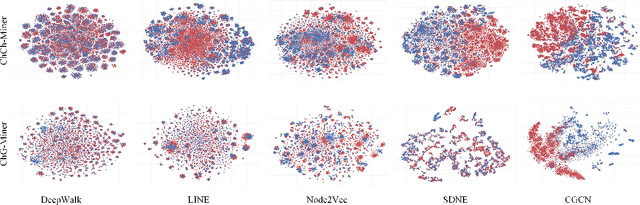
Geometric deep learning has demonstrated a great potential in non-Euclidean data analysis. The incorporation of geometric insights into learning architecture is vital to its success. Here we propose a curvature-enhanced graph convolutional network (CGCN) for biomolecular interaction prediction, for the first time. Our CGCN employs Ollivier-Ricci curvature (ORC) to characterize network local structures and to enhance the learning capability of GCNs. More specifically, ORCs are evaluated based on the local topology from node neighborhoods, and further used as weights for the feature aggregation in message-passing procedure. Our CGCN model is extensively validated on fourteen real-world bimolecular interaction networks and a series of simulated data. It has been found that our CGCN can achieve the state-of-the-art results. It outperforms all existing models, as far as we know, in thirteen out of the fourteen real-world datasets and ranks as the second in the rest one. The results from the simulated data show that our CGCN model is superior to the traditional GCN models regardless of the positive-to-negativecurvature ratios, network densities, and network sizes (when larger than 500).