 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHiPath: Hierarchical Vision-Language Alignment for Structured Pathology Report Prediction
Mar 20, 2026Pathology reports are structured, multi-granular documents encoding diagnostic conclusions, histological grades, and ancillary test results across one or more anatomical sites; yet existing pathology vision-language models (VLMs) reduce this output to a flat label or free-form text. We present HiPath, a lightweight VLM framework built on frozen UNI2 and Qwen3 backbones that treats structured report prediction as its primary training objective. Three trainable modules totalling 15M parameters address complementary aspects of the problem: a Hierarchical Patch Aggregator (HiPA) for multi-image visual encoding, Hierarchical Contrastive Learning (HiCL) for cross-modal alignment via optimal transport, and Slot-based Masked Diagnosis Prediction (Slot-MDP) for structured diagnosis generation. Trained on 749K real-world Chinese pathology cases from three hospitals, HiPath achieves 68.9% strict and 74.7% clinically acceptable accuracy with a 97.3% safety rate, outperforming all baselines under the same frozen backbone. Cross-hospital evaluation confirms generalisation with only a 3.4pp drop in strict accuracy while maintaining 97.1% safety.
Multi-DNN Inference of Sparse Models on Edge SoCs
Mar 10, 2026Modern edge applications increasingly require multi-DNN inference systems to execute tasks on heterogeneous processors, gaining performance from both concurrent execution and from matching each model to the most suited accelerator. However, existing systems support only a single model (or a few sparse variants) per task, which impedes the efficiency of this matching and results in high Service Level Objective violation rates. We introduce model stitching for multi-DNN inference systems, which creates model variants by recombining subgraphs from sparse models without re-training. We present a demonstrator system, SparseLoom, that shows model stitching can be deployed to SoCs. We show experimentally that SparseLoom reduces SLO violation rates by up to 74%, improves throughput by up to 2.31x, and lowers memory overhead by an average of 28% compared to state-of-the-art multi-DNN inference systems.
Ultra-Wideband Transmission Systems From an Energy Perspective: Which Band is Next?
Jan 08, 2026Measuring the power efficiency of the state-of-the-art OESCL-band amplifiers, we show that 1000 km OESCL-band systems can achieve 2.98x greater throughput for +48% higher energy-per-bit compared to CL-band transmission only.
Curvature-enhanced Graph Convolutional Network for Biomolecular Interaction Prediction
Jun 23, 2023
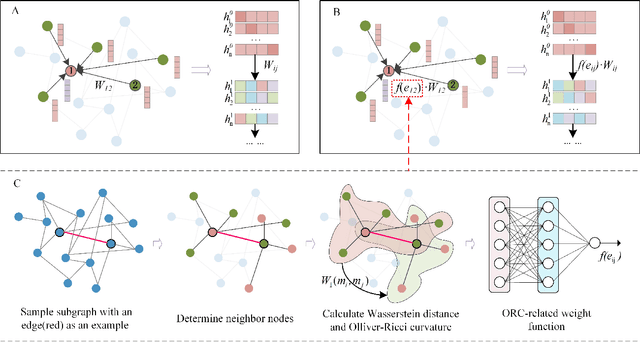
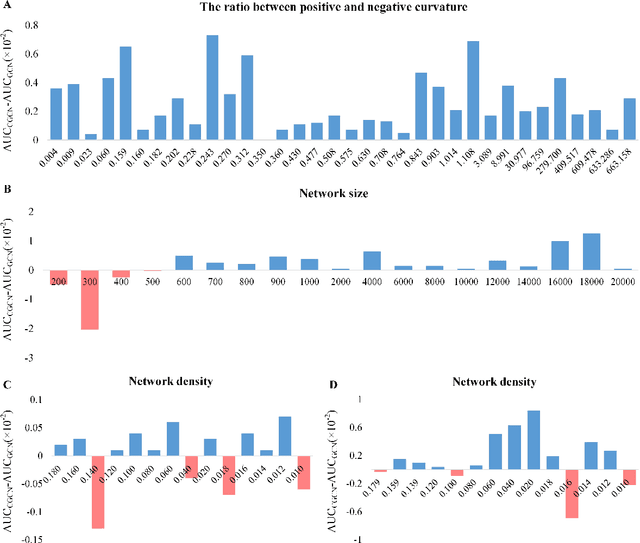
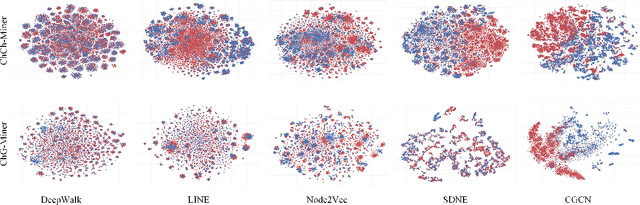
Geometric deep learning has demonstrated a great potential in non-Euclidean data analysis. The incorporation of geometric insights into learning architecture is vital to its success. Here we propose a curvature-enhanced graph convolutional network (CGCN) for biomolecular interaction prediction, for the first time. Our CGCN employs Ollivier-Ricci curvature (ORC) to characterize network local structures and to enhance the learning capability of GCNs. More specifically, ORCs are evaluated based on the local topology from node neighborhoods, and further used as weights for the feature aggregation in message-passing procedure. Our CGCN model is extensively validated on fourteen real-world bimolecular interaction networks and a series of simulated data. It has been found that our CGCN can achieve the state-of-the-art results. It outperforms all existing models, as far as we know, in thirteen out of the fourteen real-world datasets and ranks as the second in the rest one. The results from the simulated data show that our CGCN model is superior to the traditional GCN models regardless of the positive-to-negativecurvature ratios, network densities, and network sizes (when larger than 500).
Torsion Graph Neural Networks
Jun 23, 2023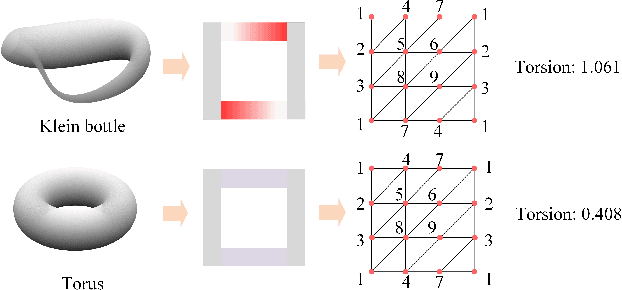
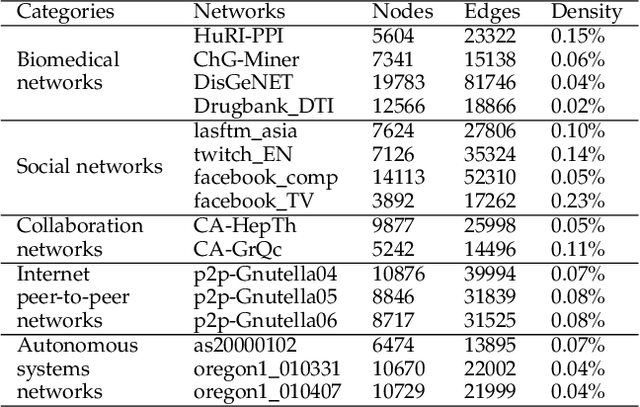
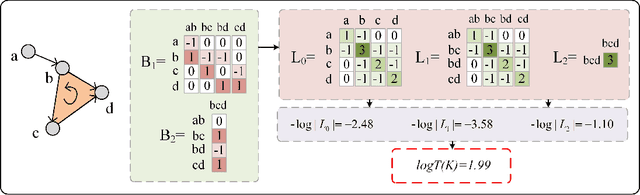
Geometric deep learning (GDL) models have demonstrated a great potential for the analysis of non-Euclidian data. They are developed to incorporate the geometric and topological information of non-Euclidian data into the end-to-end deep learning architectures. Motivated by the recent success of discrete Ricci curvature in graph neural network (GNNs), we propose TorGNN, an analytic Torsion enhanced Graph Neural Network model. The essential idea is to characterize graph local structures with an analytic torsion based weight formula. Mathematically, analytic torsion is a topological invariant that can distinguish spaces which are homotopy equivalent but not homeomorphic. In our TorGNN, for each edge, a corresponding local simplicial complex is identified, then the analytic torsion (for this local simplicial complex) is calculated, and further used as a weight (for this edge) in message-passing process. Our TorGNN model is validated on link prediction tasks from sixteen different types of networks and node classification tasks from three types of networks. It has been found that our TorGNN can achieve superior performance on both tasks, and outperform various state-of-the-art models. This demonstrates that analytic torsion is a highly efficient topological invariant in the characterization of graph structures and can significantly boost the performance of GNNs.
Molecular geometric deep learning
Jun 22, 2023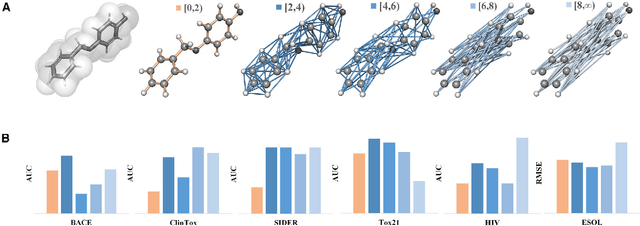
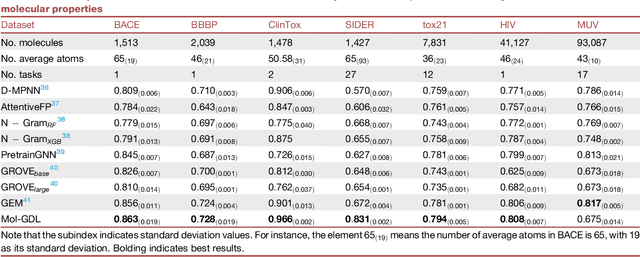
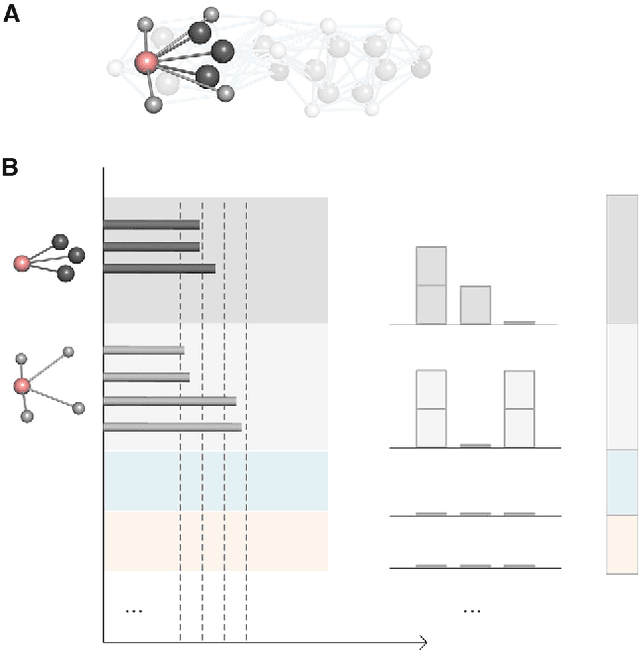
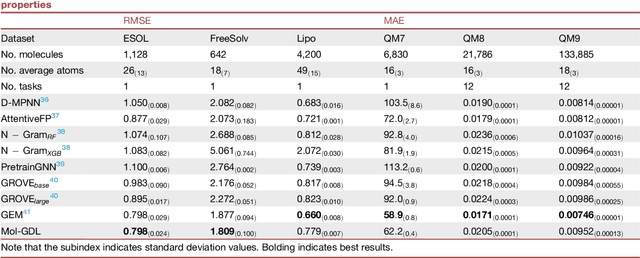
Geometric deep learning (GDL) has demonstrated huge power and enormous potential in molecular data analysis. However, a great challenge still remains for highly efficient molecular representations. Currently, covalent-bond-based molecular graphs are the de facto standard for representing molecular topology at the atomic level. Here we demonstrate, for the first time, that molecular graphs constructed only from non-covalent bonds can achieve similar or even better results than covalent-bond-based models in molecular property prediction. This demonstrates the great potential of novel molecular representations beyond the de facto standard of covalent-bond-based molecular graphs. Based on the finding, we propose molecular geometric deep learning (Mol-GDL). The essential idea is to incorporate a more general molecular representation into GDL models. In our Mol-GDL, molecular topology is modeled as a series of molecular graphs, each focusing on a different scale of atomic interactions. In this way, both covalent interactions and non-covalent interactions are incorporated into the molecular representation on an equal footing. We systematically test Mol-GDL on fourteen commonly-used benchmark datasets. The results show that our Mol-GDL can achieve a better performance than state-of-the-art (SOTA) methods. Source code and data are available at https://github.com/CS-BIO/Mol-GDL.
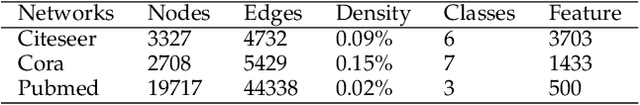
iEnhancer-ELM: Improve Enhancer Identification by Extracting Multi-scale Contextual Information based on Enhancer Language Models
Dec 03, 2022Motivation: Enhancers are important cis-regulatory elements that regulate a wide range of biological functions and enhance the transcription of target genes. Although many state-of-the-art computational methods have been proposed in order to efficiently identify enhancers, learning globally contextual features is still one of the challenges for computational methods. Regarding the similarities between biological sequences and natural language sentences, the novel BERT-based language techniques have been applied to extracting complex contextual features in various computational biology tasks such as protein function/structure prediction. To speed up the research on enhancer identification, it is urgent to construct a BERT-based enhancer language model. Results: In this paper, we propose a multi-scale enhancer identification method (iEnhancer-ELM) based on enhancer language models, which treat enhancer sequences as natural language sentences that are composed of k-mer nucleotides. iEnhancer-ELM can extract contextual information of multi-scale k-mers with positions from raw enhancer sequences. Benefiting from the complementary information of k-mers in multi-scale, we ensemble four iEnhancer-ELM models for improving enhancer identification. The benchmark comparisons show that our model outperforms state-of-the-art methods. By the interpretable attention mechanism, we finds 30 biological patterns, where 40% (12/30) are verified by a widely used motif tool (STREME) and a popular dataset (JASPAR), demonstrating our model has a potential ability to reveal the biological mechanism of enhancer. Availability: The source code are available at https://github.com/chen-bioinfo/iEnhancer-ELM Contact: junjiechen@hit.edu.cn and junjie.chen.hit@gmail.com; Supplementary information: Supplementary data are available at Bioinformatics online.