 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeICON: Intent-Context Coupling for Efficient Multi-Turn Jailbreak Attack
Jan 28, 2026Multi-turn jailbreak attacks have emerged as a critical threat to Large Language Models (LLMs), bypassing safety mechanisms by progressively constructing adversarial contexts from scratch and incrementally refining prompts. However, existing methods suffer from the inefficiency of incremental context construction that requires step-by-step LLM interaction, and often stagnate in suboptimal regions due to surface-level optimization. In this paper, we characterize the Intent-Context Coupling phenomenon, revealing that LLM safety constraints are significantly relaxed when a malicious intent is coupled with a semantically congruent context pattern. Driven by this insight, we propose ICON, an automated multi-turn jailbreak framework that efficiently constructs an authoritative-style context via prior-guided semantic routing. Specifically, ICON first routes the malicious intent to a congruent context pattern (e.g., Scientific Research) and instantiates it into an attack prompt sequence. This sequence progressively builds the authoritative-style context and ultimately elicits prohibited content. In addition, ICON incorporates a Hierarchical Optimization Strategy that combines local prompt refinement with global context switching, preventing the attack from stagnating in ineffective contexts. Experimental results across eight SOTA LLMs demonstrate the effectiveness of ICON, achieving a state-of-the-art average Attack Success Rate (ASR) of 97.1\%. Code is available at https://github.com/xwlin-roy/ICON.
What Kind of Visual Tokens Do We Need? Training-free Visual Token Pruning for Multi-modal Large Language Models from the Perspective of Graph
Jan 04, 2025



Recent Multimodal Large Language Models(MLLMs) often use a large number of visual tokens to compensate their visual shortcoming, leading to excessive computation and obvious visual redundancy. In this paper, we investigate what kind of visual tokens are needed for MLLMs, and reveal that both foreground and background tokens are critical for MLLMs given the varying difficulties of examples. Based on this observation, we propose a graph-based method towards training-free visual token pruning, termed G-Prune.In particular, G-Prune regards visual tokens as nodes, and construct their connections based on their semantic similarities. Afterwards, the information flow is propagated via weighted links, and the most important tokens after iterations are kept for MLLMs, which can be front or background.To validate G-Prune, we apply it to a recent MLLM called LLaVA-NeXT, and conduct extensive experiments on a set of benchmarks.The experiment results show that G-Prune can greatly reduce computation overhead while retaining high performance on both coarse- and fine-grained tasks. For instance, G-Prune can reduce 63.57\% FLOPs of LLaVA-NeXT on VQA2.0 and TextVQA with only 0.95\% and 2.34\% accuracy drops, respectively.
Accelerating Multimodal Large Language Models via Dynamic Visual-Token Exit and the Empirical Findings
Nov 29, 2024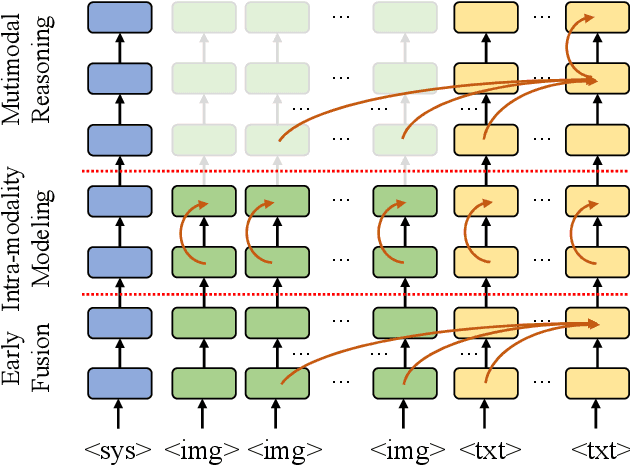


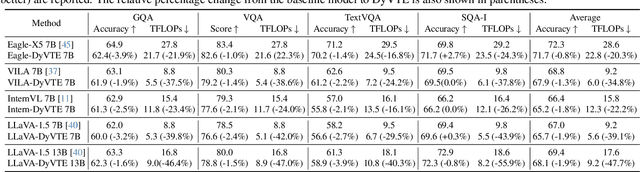
The excessive use of visual tokens in existing Multimoal Large Language Models (MLLMs) often exhibits obvious redundancy and brings in prohibitively expensive computation. To gain insights into this problem, we first conduct extensive empirical studies on the attention behaviors of MLLMs, and summarize three main inference stages in MLLMs: (i) Early fusion between tokens is first accomplished quickly. (ii) Intra-modality modeling then comes to play. (iii) Multimodal reasoning} resumes and lasts until the end of inference. In particular, we reveal that visual tokens will stop contributing to reasoning when the text tokens receive enough image information, yielding obvious visual redundancy. Based on these generalized observations, we propose a simple yet effective method to improve the efficiency of MLLMs, termed dynamic visual-token exit (DyVTE). DyVTE uses lightweight hyper-networks to perceive the text token status and decide the removal of all visual tokens after a certain layer, thereby addressing the observed visual redundancy. To validate VTE, we apply it to a set of MLLMs, including LLaVA, VILA, Eagle and InternVL, and conduct extensive experiments on a bunch of benchmarks. The experiment results not only show the effectiveness of our VTE in improving MLLMs' efficiency, but also yield the general modeling patterns of MLLMs, well facilitating the in-depth understanding of MLLMs. Our code is anonymously released at https://github.com/DoubtedSteam/DyVTE.
Fit and Prune: Fast and Training-free Visual Token Pruning for Multi-modal Large Language Models
Sep 16, 2024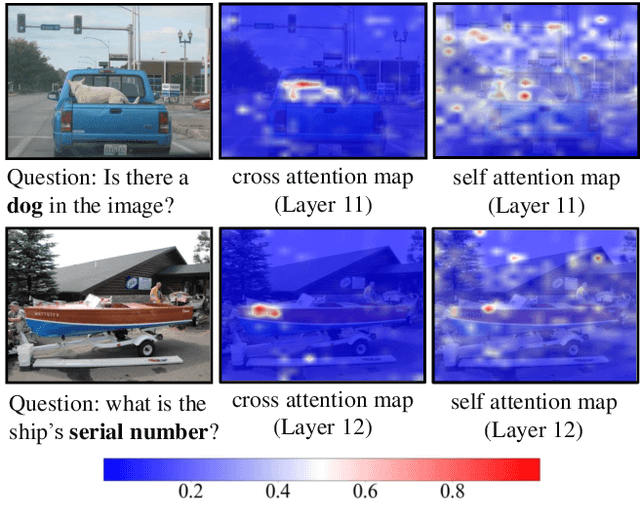



Recent progress in Multimodal Large Language Models(MLLMs) often use large image tokens to compensate the visual shortcoming of MLLMs, which not only exhibits obvious redundancy but also greatly exacerbates the already high computation. Token pruning is an effective solution for speeding up MLLMs, but when and how to drop tokens still remains a challenge. In this paper, we propose a novel and training-free approach for the effective visual token pruning of MLLMs, termed FitPrune, which can quickly produce a complete pruning recipe for MLLMs according to a pre-defined budget. Specifically, FitPrune considers token pruning as a statistical problem of MLLM and its objective is to find out an optimal pruning scheme that can minimize the divergence of the attention distributions before and after pruning. In practice, FitPrune can be quickly accomplished based on the attention statistics from a small batch of inference data, avoiding the expensive trials of MLLMs. According to the pruning recipe, an MLLM can directly remove the redundant visual tokens of different examples during inference. To validate FitPrune, we apply it to a set of recent MLLMs, including LLaVA-1.5, LLaVA-HR and LLaVA-NEXT, and conduct extensive experiments on a set of benchmarks. The experimental results show that our FitPrune can not only reduce the computational complexity to a large extent, while retaining high performance, e.g., -54.9% FLOPs for LLaVA-NEXT with only 0.5% accuracy drop. Notably, the pruning recipe can be obtained in about 5 minutes. Our code is available at https://github.com/ywh187/FitPrune.
iEnhancer-ELM: Improve Enhancer Identification by Extracting Multi-scale Contextual Information based on Enhancer Language Models
Dec 03, 2022Motivation: Enhancers are important cis-regulatory elements that regulate a wide range of biological functions and enhance the transcription of target genes. Although many state-of-the-art computational methods have been proposed in order to efficiently identify enhancers, learning globally contextual features is still one of the challenges for computational methods. Regarding the similarities between biological sequences and natural language sentences, the novel BERT-based language techniques have been applied to extracting complex contextual features in various computational biology tasks such as protein function/structure prediction. To speed up the research on enhancer identification, it is urgent to construct a BERT-based enhancer language model. Results: In this paper, we propose a multi-scale enhancer identification method (iEnhancer-ELM) based on enhancer language models, which treat enhancer sequences as natural language sentences that are composed of k-mer nucleotides. iEnhancer-ELM can extract contextual information of multi-scale k-mers with positions from raw enhancer sequences. Benefiting from the complementary information of k-mers in multi-scale, we ensemble four iEnhancer-ELM models for improving enhancer identification. The benchmark comparisons show that our model outperforms state-of-the-art methods. By the interpretable attention mechanism, we finds 30 biological patterns, where 40% (12/30) are verified by a widely used motif tool (STREME) and a popular dataset (JASPAR), demonstrating our model has a potential ability to reveal the biological mechanism of enhancer. Availability: The source code are available at https://github.com/chen-bioinfo/iEnhancer-ELM Contact: junjiechen@hit.edu.cn and junjie.chen.hit@gmail.com; Supplementary information: Supplementary data are available at Bioinformatics online.
Generative Data Augmentation for Non-IID Problem in Decentralized Clinical Machine Learning
Dec 02, 2022



Swarm learning (SL) is an emerging promising decentralized machine learning paradigm and has achieved high performance in clinical applications. SL solves the problem of a central structure in federated learning by combining edge computing and blockchain-based peer-to-peer network. While there are promising results in the assumption of the independent and identically distributed (IID) data across participants, SL suffers from performance degradation as the degree of the non-IID data increases. To address this problem, we propose a generative augmentation framework in swarm learning called SL-GAN, which augments the non-IID data by generating the synthetic data from participants. SL-GAN trains generators and discriminators locally, and periodically aggregation via a randomly elected coordinator in SL network. Under the standard assumptions, we theoretically prove the convergence of SL-GAN using stochastic approximations. Experimental results demonstrate that SL-GAN outperforms state-of-art methods on three real world clinical datasets including Tuberculosis, Leukemia, COVID-19.