 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular geometric deep learning
Paper and Code
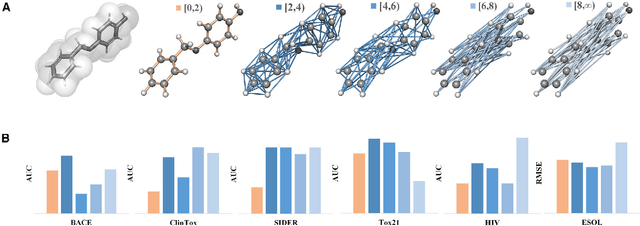
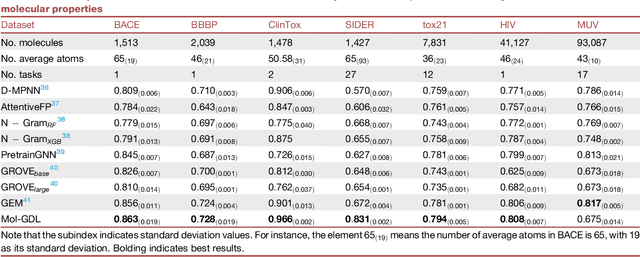
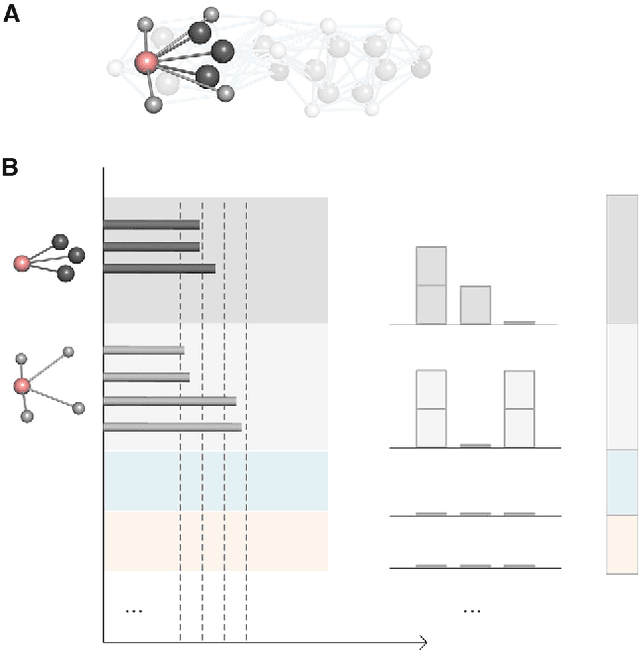
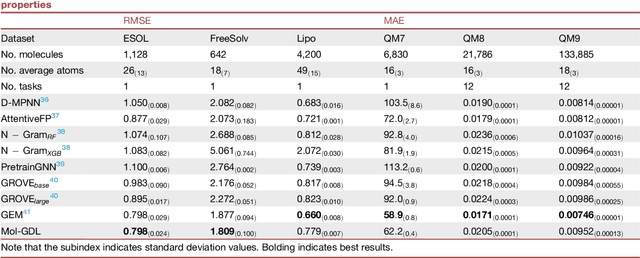
Geometric deep learning (GDL) has demonstrated huge power and enormous potential in molecular data analysis. However, a great challenge still remains for highly efficient molecular representations. Currently, covalent-bond-based molecular graphs are the de facto standard for representing molecular topology at the atomic level. Here we demonstrate, for the first time, that molecular graphs constructed only from non-covalent bonds can achieve similar or even better results than covalent-bond-based models in molecular property prediction. This demonstrates the great potential of novel molecular representations beyond the de facto standard of covalent-bond-based molecular graphs. Based on the finding, we propose molecular geometric deep learning (Mol-GDL). The essential idea is to incorporate a more general molecular representation into GDL models. In our Mol-GDL, molecular topology is modeled as a series of molecular graphs, each focusing on a different scale of atomic interactions. In this way, both covalent interactions and non-covalent interactions are incorporated into the molecular representation on an equal footing. We systematically test Mol-GDL on fourteen commonly-used benchmark datasets. The results show that our Mol-GDL can achieve a better performance than state-of-the-art (SOTA) methods. Source code and data are available at https://github.com/CS-BIO/Mol-GDL.