 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards a Rigorous Evaluation of Time-series Anomaly Detection
Sep 11, 2021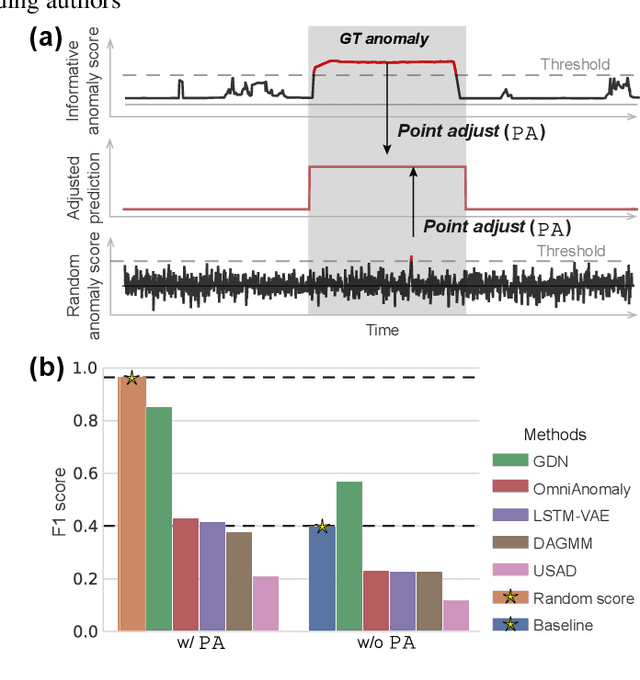
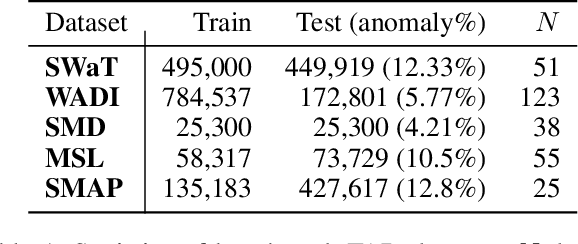
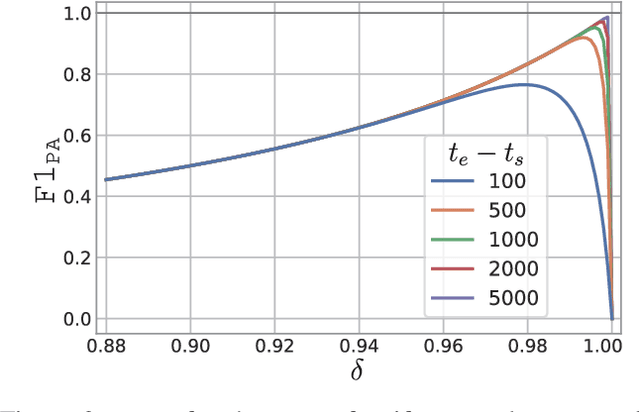
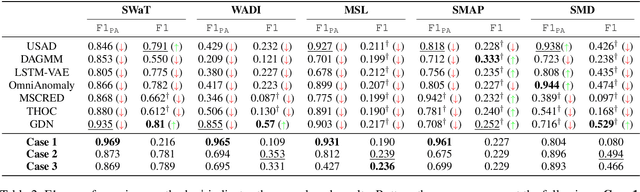
In recent years, proposed studies on time-series anomaly detection (TAD) report high F1 scores on benchmark TAD datasets, giving the impression of clear improvements. However, most studies apply a peculiar evaluation protocol called point adjustment (PA) before scoring. In this paper, we theoretically and experimentally reveal that the PA protocol has a great possibility of overestimating the detection performance; that is, even a random anomaly score can easily turn into a state-of-the-art TAD method. Therefore, the comparison of TAD methods with F1 scores after the PA protocol can lead to misguided rankings. Furthermore, we question the potential of existing TAD methods by showing that an untrained model obtains comparable detection performance to the existing methods even without PA. Based on our findings, we propose a new baseline and an evaluation protocol. We expect that our study will help a rigorous evaluation of TAD and lead to further improvement in future researches.
TargetNet: Functional microRNA Target Prediction with Deep Neural Networks
Jul 23, 2021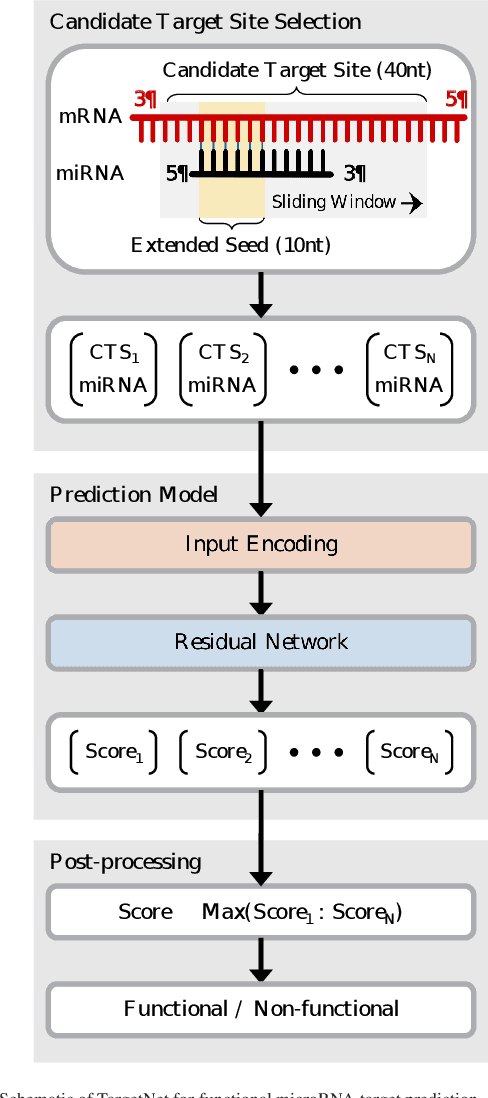
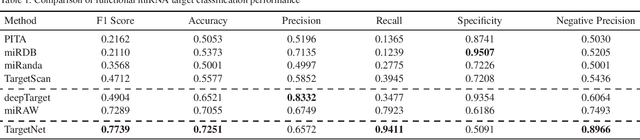
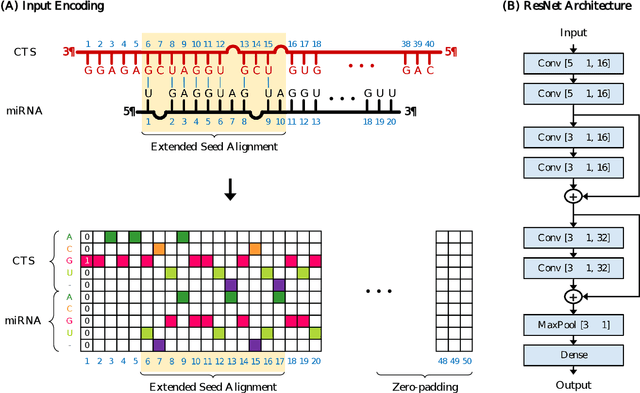
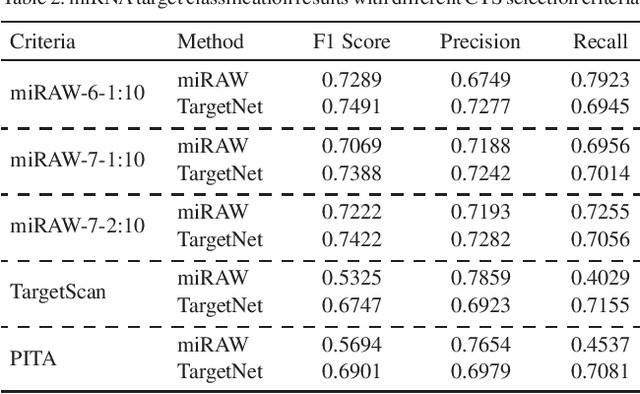
MicroRNAs (miRNAs) play pivotal roles in gene expression regulation by binding to target sites of messenger RNAs (mRNAs). While identifying functional targets of miRNAs is of utmost importance, their prediction remains a great challenge. Previous computational algorithms have major limitations. They use conservative candidate target site (CTS) selection criteria mainly focusing on canonical site types, rely on laborious and time-consuming manual feature extraction, and do not fully capitalize on the information underlying miRNA-CTS interactions. In this paper, we introduce TargetNet, a novel deep learning-based algorithm for functional miRNA target prediction. To address the limitations of previous approaches, TargetNet has three key components: (1) relaxed CTS selection criteria accommodating irregularities in the seed region, (2) a novel miRNA-CTS sequence encoding scheme incorporating extended seed region alignments, and (3) a deep residual network-based prediction model. The proposed model was trained with miRNA-CTS pair datasets and evaluated with miRNA-mRNA pair datasets. TargetNet advances the previous state-of-the-art algorithms used in functional miRNA target classification. Furthermore, it demonstrates great potential for distinguishing high-functional miRNA targets.
Geometry-aware Transformer for molecular property prediction
Jun 29, 2021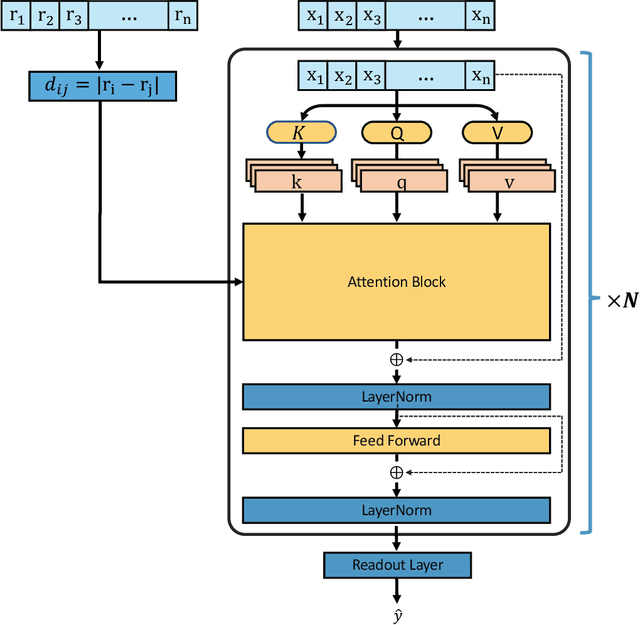
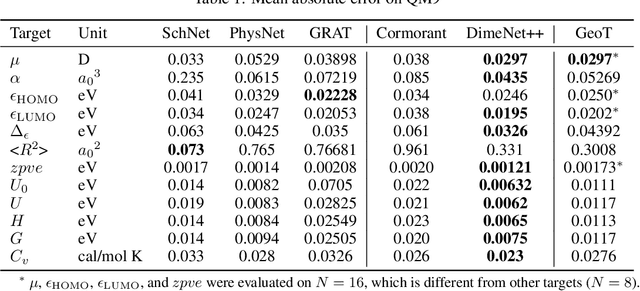
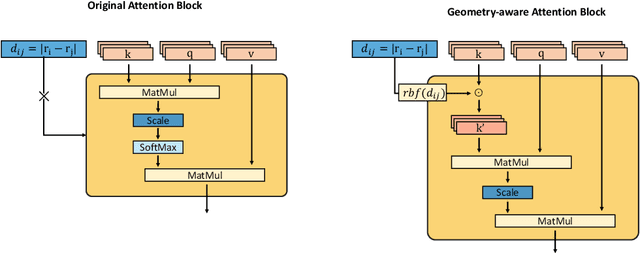
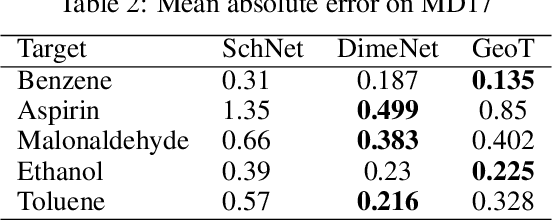
Recently, graph neural networks (GNNs) have achieved remarkable performances for quantum mechanical problems. However, a graph convolution can only cover a localized region, and cannot capture long-range interactions of atoms. This behavior is contrary to theoretical interatomic potentials, which is a fundamental limitation of the spatial based GNNs. In this work, we propose a novel attention-based framework for molecular property prediction tasks. We represent a molecular conformation as a discrete atomic sequence combined by atom-atom distance attributes, named Geometry-aware Transformer (GeoT). In particular, we adopt a Transformer architecture, which has been widely used for sequential data. Our proposed model trains sequential representations of molecular graphs based on globally constructed attentions, maintaining all spatial arrangements of atom pairs. Our method does not suffer from cost intensive computations, such as angle calculations. The experimental results on several public benchmarks and visualization maps verified that keeping the long-range interatomic attributes can significantly improve the model predictability.
Flexible dual-branched message passing neural network for quantum mechanical property prediction with molecular conformation
Jun 14, 2021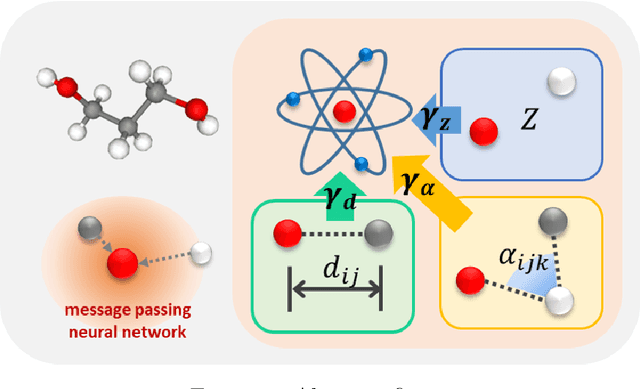
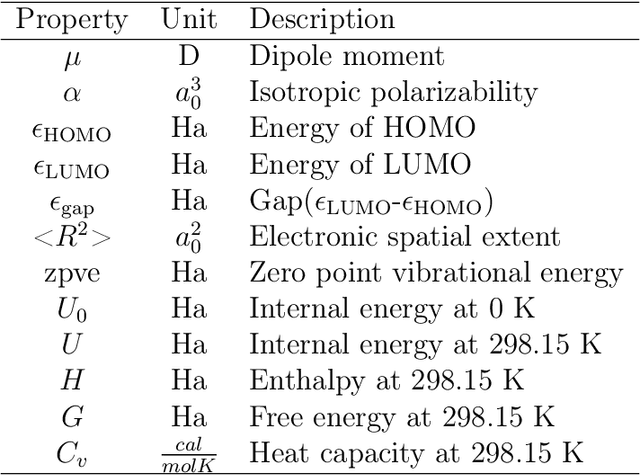
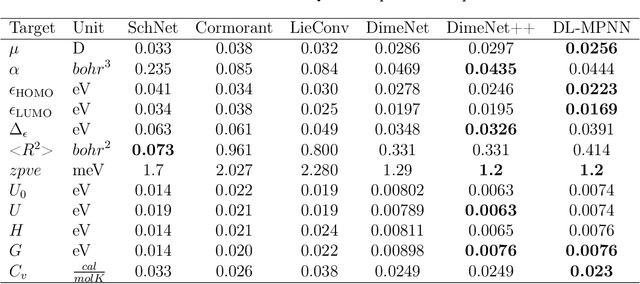
A molecule is a complex of heterogeneous components, and the spatial arrangements of these components determine the whole molecular properties and characteristics. With the advent of deep learning in computational chemistry, several studies have focused on how to predict molecular properties based on molecular configurations. Message passing neural network provides an effective framework for capturing molecular geometric features with the perspective of a molecule as a graph. However, most of these studies assumed that all heterogeneous molecular features, such as atomic charge, bond length, or other geometric features always contribute equivalently to the target prediction, regardless of the task type. In this study, we propose a dual-branched neural network for molecular property prediction based on message-passing framework. Our model learns heterogeneous molecular features with different scales, which are trained flexibly according to each prediction target. In addition, we introduce a discrete branch to learn single atom features without local aggregation, apart from message-passing steps. We verify that this novel structure can improve the model performance with faster convergence in most targets. The proposed model outperforms other recent models with sparser representations. Our experimental results indicate that in the chemical property prediction tasks, the diverse chemical nature of targets should be carefully considered for both model performance and generalizability.
DNA Steganalysis Using Deep Recurrent Neural Networks
Oct 05, 2018

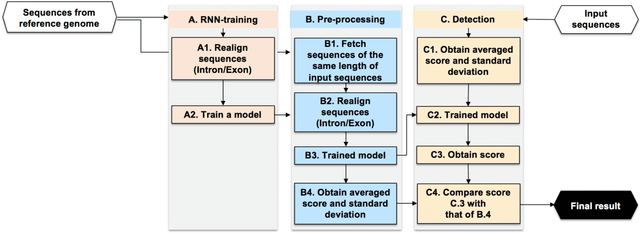
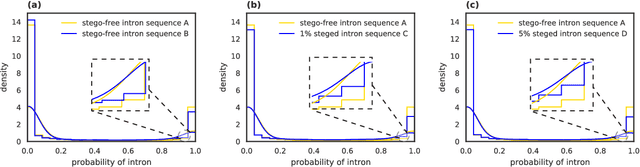
Recent advances in next-generation sequencing technologies have facilitated the use of deoxyribonucleic acid (DNA) as a novel covert channels in steganography. There are various methods that exist in other domains to detect hidden messages in conventional covert channels. However, they have not been applied to DNA steganography. The current most common detection approaches, namely frequency analysis-based methods, often overlook important signals when directly applied to DNA steganography because those methods depend on the distribution of the number of sequence characters. To address this limitation, we propose a general sequence learning-based DNA steganalysis framework. The proposed approach learns the intrinsic distribution of coding and non-coding sequences and detects hidden messages by exploiting distribution variations after hiding these messages. Using deep recurrent neural networks (RNNs), our framework identifies the distribution variations by using the classification score to predict whether a sequence is to be a coding or non-coding sequence. We compare our proposed method to various existing methods and biological sequence analysis methods implemented on top of our framework. According to our experimental results, our approach delivers a robust detection performance compared to other tools.
Deep Learning on Key Performance Indicators for Predictive Maintenance in SAP HANA
Apr 16, 2018
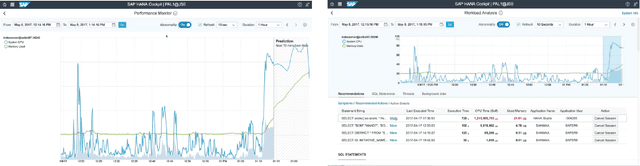
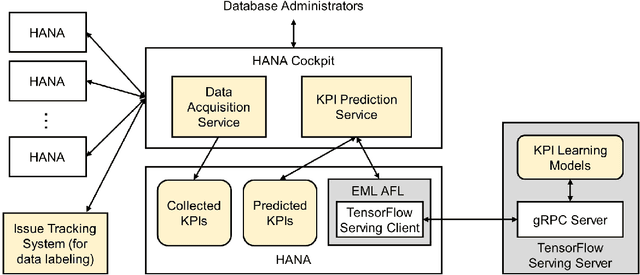

With a new era of cloud and big data, Database Management Systems (DBMSs) have become more crucial in numerous enterprise business applications in all the industries. Accordingly, the importance of their proactive and preventive maintenance has also increased. However, detecting problems by predefined rules or stochastic modeling has limitations, particularly when analyzing the data on high-dimensional Key Performance Indicators (KPIs) from a DBMS. In recent years, Deep Learning (DL) has opened new opportunities for this complex analysis. In this paper, we present two complementary DL approaches to detect anomalies in SAP HANA. A temporal learning approach is used to detect abnormal patterns based on unlabeled historical data, whereas a spatial learning approach is used to classify known anomalies based on labeled data. We implement a system in SAP HANA integrated with Google TensorFlow. The experimental results with real-world data confirm the effectiveness of the system and models.
Neural Universal Discrete Denoiser
Aug 24, 2016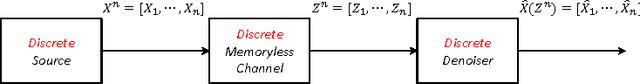
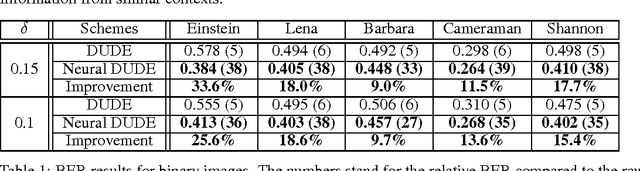
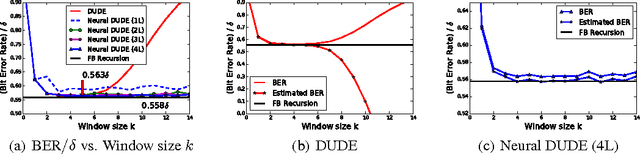
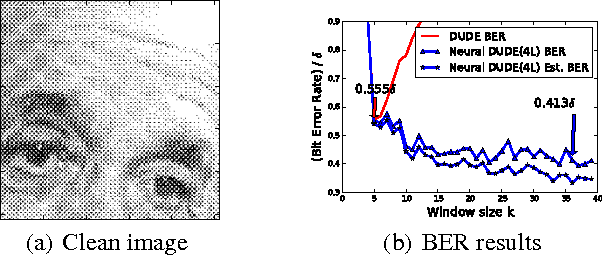
We present a new framework of applying deep neural networks (DNN) to devise a universal discrete denoiser. Unlike other approaches that utilize supervised learning for denoising, we do not require any additional training data. In such setting, while the ground-truth label, i.e., the clean data, is not available, we devise "pseudo-labels" and a novel objective function such that DNN can be trained in a same way as supervised learning to become a discrete denoiser. We experimentally show that our resulting algorithm, dubbed as Neural DUDE, significantly outperforms the previous state-of-the-art in several applications with a systematic rule of choosing the hyperparameter, which is an attractive feature in practice.
deepTarget: End-to-end Learning Framework for microRNA Target Prediction using Deep Recurrent Neural Networks
Aug 19, 2016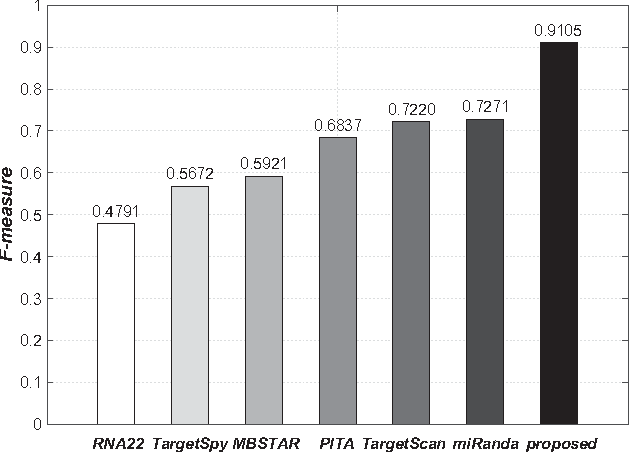
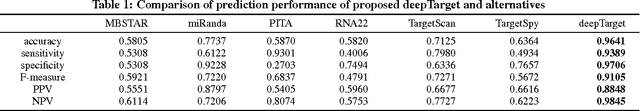
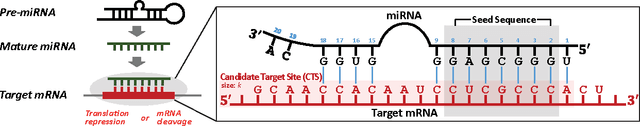
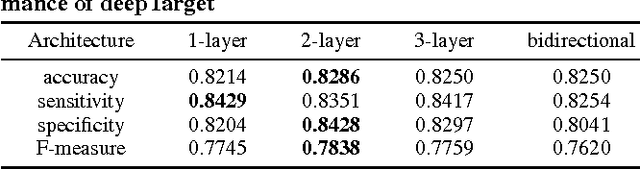
MicroRNAs (miRNAs) are short sequences of ribonucleic acids that control the expression of target messenger RNAs (mRNAs) by binding them. Robust prediction of miRNA-mRNA pairs is of utmost importance in deciphering gene regulations but has been challenging because of high false positive rates, despite a deluge of computational tools that normally require laborious manual feature extraction. This paper presents an end-to-end machine learning framework for miRNA target prediction. Leveraged by deep recurrent neural networks-based auto-encoding and sequence-sequence interaction learning, our approach not only delivers an unprecedented level of accuracy but also eliminates the need for manual feature extraction. The performance gap between the proposed method and existing alternatives is substantial (over 25% increase in F-measure), and deepTarget delivers a quantum leap in the long-standing challenge of robust miRNA target prediction.
Deep Learning in Bioinformatics
Jun 19, 2016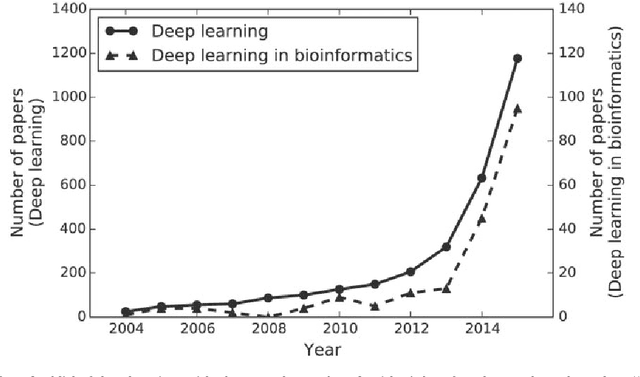
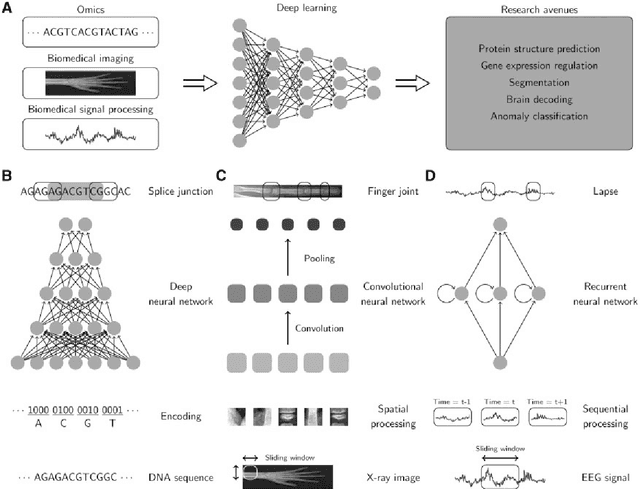
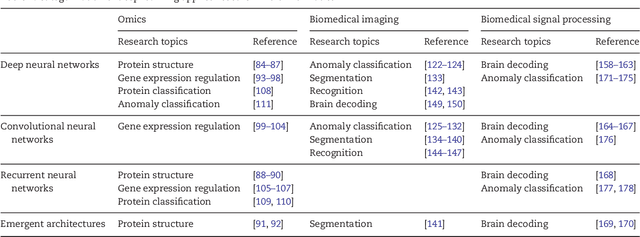
In the era of big data, transformation of biomedical big data into valuable knowledge has been one of the most important challenges in bioinformatics. Deep learning has advanced rapidly since the early 2000s and now demonstrates state-of-the-art performance in various fields. Accordingly, application of deep learning in bioinformatics to gain insight from data has been emphasized in both academia and industry. Here, we review deep learning in bioinformatics, presenting examples of current research. To provide a useful and comprehensive perspective, we categorize research both by the bioinformatics domain (i.e., omics, biomedical imaging, biomedical signal processing) and deep learning architecture (i.e., deep neural networks, convolutional neural networks, recurrent neural networks, emergent architectures) and present brief descriptions of each study. Additionally, we discuss theoretical and practical issues of deep learning in bioinformatics and suggest future research directions. We believe that this review will provide valuable insights and serve as a starting point for researchers to apply deep learning approaches in their bioinformatics studies.
DNA-Level Splice Junction Prediction using Deep Recurrent Neural Networks
Dec 16, 2015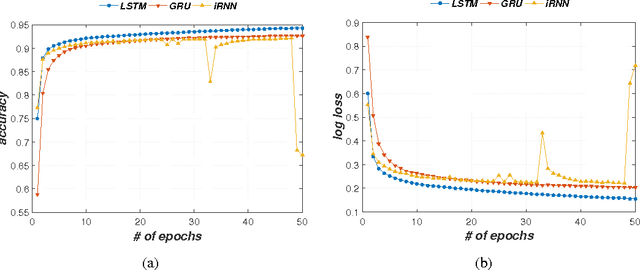
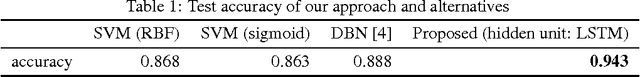
A eukaryotic gene consists of multiple exons (protein coding regions) and introns (non-coding regions), and a splice junction refers to the boundary between a pair of exon and intron. Precise identification of spice junctions on a gene is important for deciphering its primary structure, function, and interaction. Experimental techniques for determining exon/intron boundaries include RNA-seq, which is often accompanied by computational approaches. Canonical splicing signals are known, but computational junction prediction still remains challenging because of a large number of false positives and other complications. In this paper, we exploit deep recurrent neural networks (RNNs) to model DNA sequences and to detect splice junctions thereon. We test various RNN units and architectures including long short-term memory units, gated recurrent units, and recently proposed iRNN for in-depth design space exploration. According to our experimental results, the proposed approach significantly outperforms not only conventional machine learning-based methods but also a recent state-of-the-art deep belief network-based technique in terms of prediction accuracy.