 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNeural Polarization: Toward Electron Density for Molecules by Extending Equivariant Networks
Jun 01, 2024



Recent SO(3)-equivariant models embedded a molecule as a set of single atoms fixed in the three-dimensional space, which is analogous to a ball-and-stick view. This perspective provides a concise view of atom arrangements, however, the surrounding electron density cannot be represented and its polarization effects may be underestimated. To overcome this limitation, we propose \textit{Neural Polarization}, a novel method extending equivariant network by embedding each atom as a pair of fixed and moving points. Motivated by density functional theory, Neural Polarization represents molecules as a space-filling view which includes an electron density, in contrast with a ball-and-stick view. Neural Polarization can flexibly be applied to most type of existing equivariant models. We showed that Neural Polarization can improve prediction performances of existing models over a wide range of targets. Finally, we verified that our method can improve the expressiveness and equivariance in terms of mathematical aspects.
Geometry-aware Transformer for molecular property prediction
Jun 29, 2021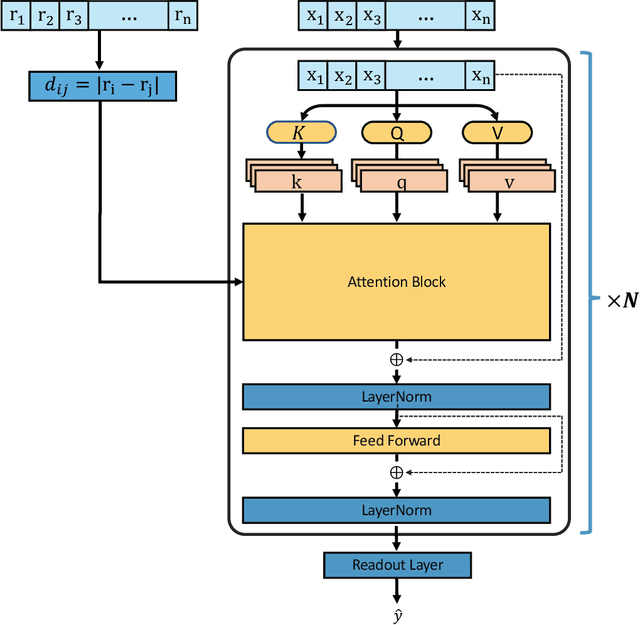
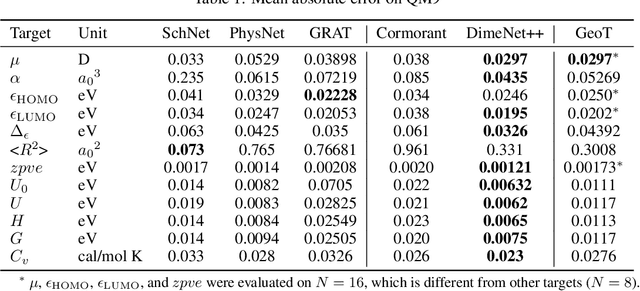
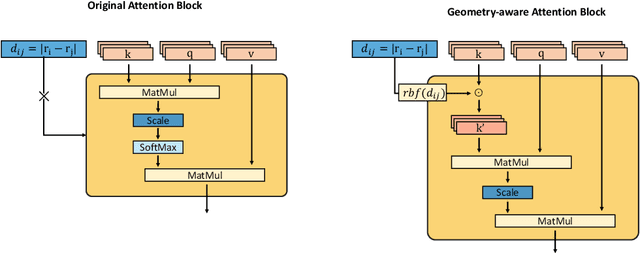
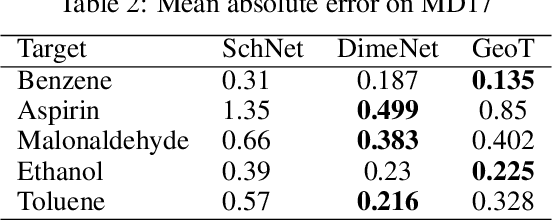
Recently, graph neural networks (GNNs) have achieved remarkable performances for quantum mechanical problems. However, a graph convolution can only cover a localized region, and cannot capture long-range interactions of atoms. This behavior is contrary to theoretical interatomic potentials, which is a fundamental limitation of the spatial based GNNs. In this work, we propose a novel attention-based framework for molecular property prediction tasks. We represent a molecular conformation as a discrete atomic sequence combined by atom-atom distance attributes, named Geometry-aware Transformer (GeoT). In particular, we adopt a Transformer architecture, which has been widely used for sequential data. Our proposed model trains sequential representations of molecular graphs based on globally constructed attentions, maintaining all spatial arrangements of atom pairs. Our method does not suffer from cost intensive computations, such as angle calculations. The experimental results on several public benchmarks and visualization maps verified that keeping the long-range interatomic attributes can significantly improve the model predictability.
Flexible dual-branched message passing neural network for quantum mechanical property prediction with molecular conformation
Jun 14, 2021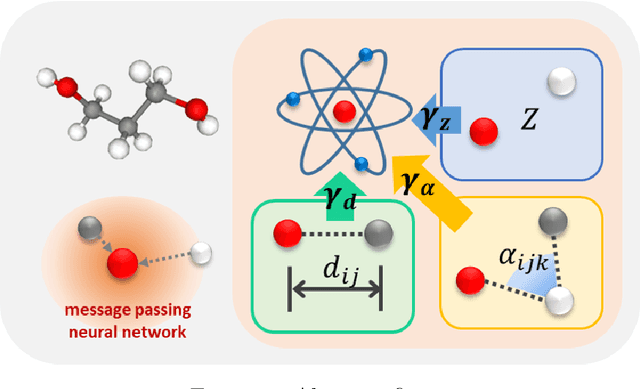
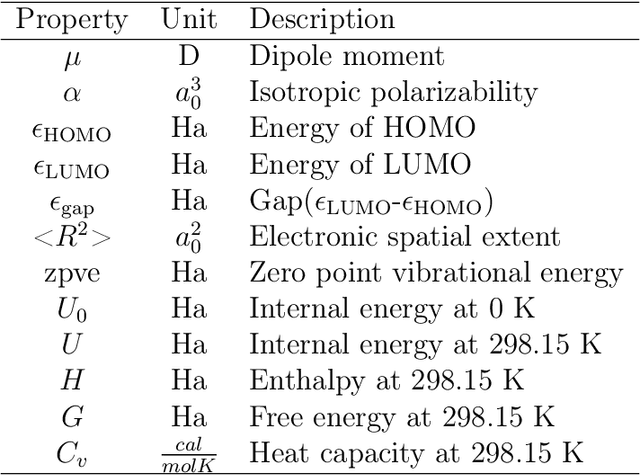
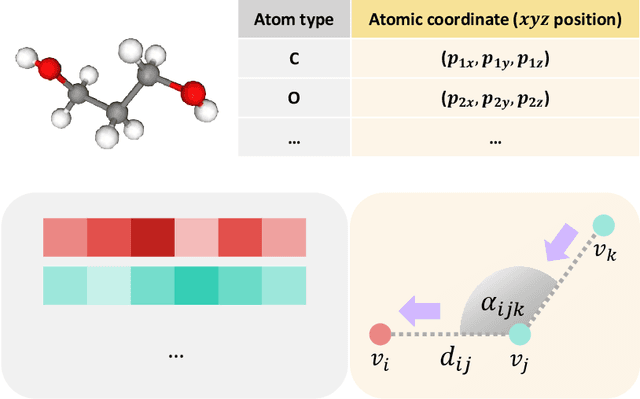
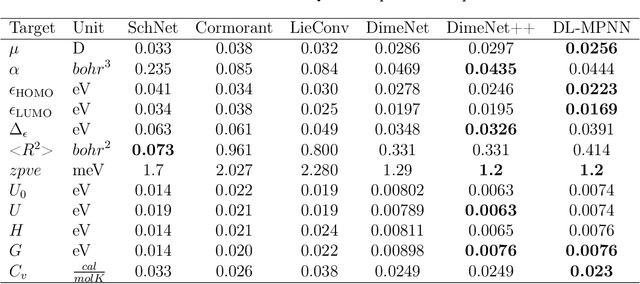
A molecule is a complex of heterogeneous components, and the spatial arrangements of these components determine the whole molecular properties and characteristics. With the advent of deep learning in computational chemistry, several studies have focused on how to predict molecular properties based on molecular configurations. Message passing neural network provides an effective framework for capturing molecular geometric features with the perspective of a molecule as a graph. However, most of these studies assumed that all heterogeneous molecular features, such as atomic charge, bond length, or other geometric features always contribute equivalently to the target prediction, regardless of the task type. In this study, we propose a dual-branched neural network for molecular property prediction based on message-passing framework. Our model learns heterogeneous molecular features with different scales, which are trained flexibly according to each prediction target. In addition, we introduce a discrete branch to learn single atom features without local aggregation, apart from message-passing steps. We verify that this novel structure can improve the model performance with faster convergence in most targets. The proposed model outperforms other recent models with sparser representations. Our experimental results indicate that in the chemical property prediction tasks, the diverse chemical nature of targets should be carefully considered for both model performance and generalizability.