 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024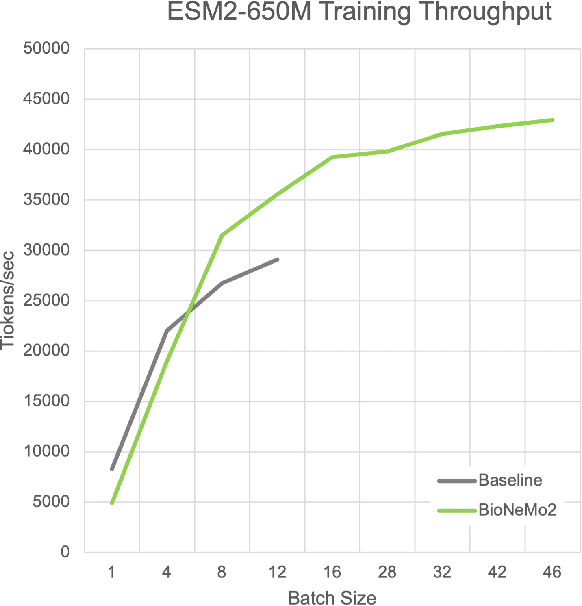
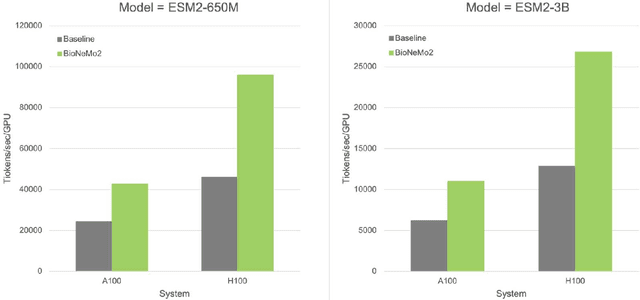
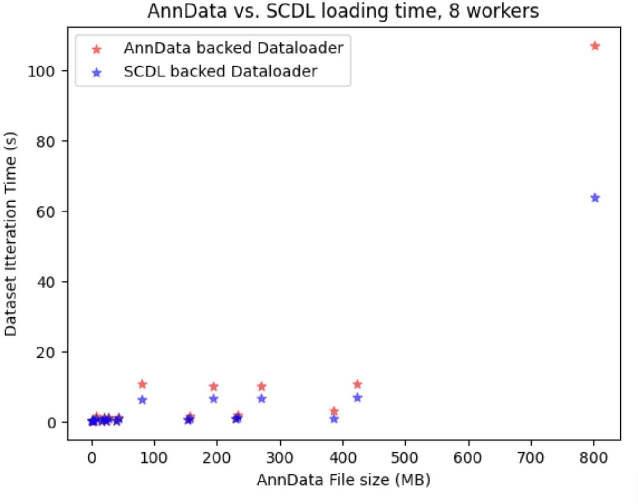
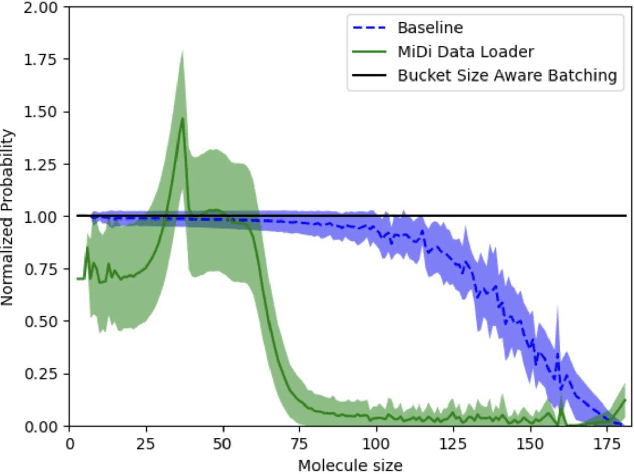
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
Ankh: Optimized Protein Language Model Unlocks General-Purpose Modelling
Jan 16, 2023As opposed to scaling-up protein language models (PLMs), we seek improving performance via protein-specific optimization. Although the proportionality between the language model size and the richness of its learned representations is validated, we prioritize accessibility and pursue a path of data-efficient, cost-reduced, and knowledge-guided optimization. Through over twenty experiments ranging from masking, architecture, and pre-training data, we derive insights from protein-specific experimentation into building a model that interprets the language of life, optimally. We present Ankh, the first general-purpose PLM trained on Google's TPU-v4 surpassing the state-of-the-art performance with fewer parameters (<10% for pre-training, <7% for inference, and <30% for the embedding dimension). We provide a representative range of structure and function benchmarks where Ankh excels. We further provide a protein variant generation analysis on High-N and One-N input data scales where Ankh succeeds in learning protein evolutionary conservation-mutation trends and introducing functional diversity while retaining key structural-functional characteristics. We dedicate our work to promoting accessibility to research innovation via attainable resources.
CodeTrans: Towards Cracking the Language of Silicone's Code Through Self-Supervised Deep Learning and High Performance Computing
Apr 06, 2021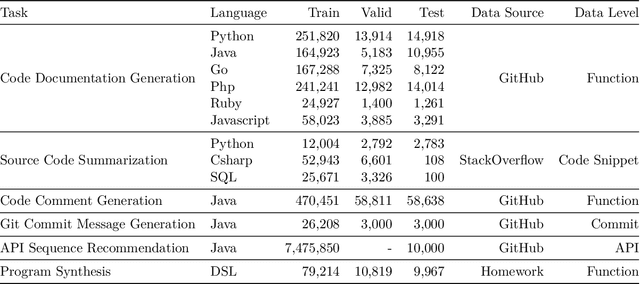
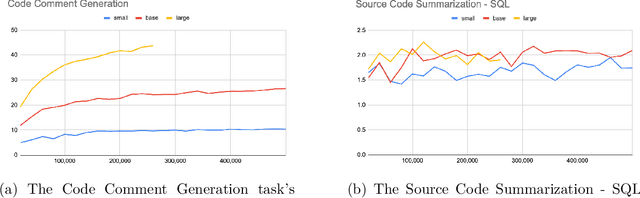
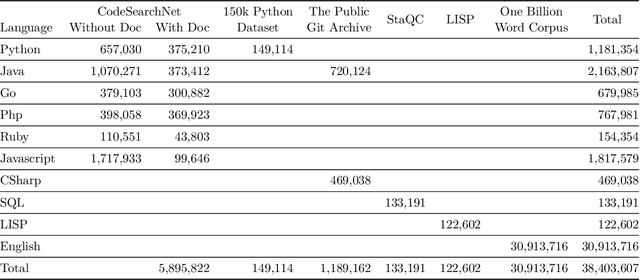
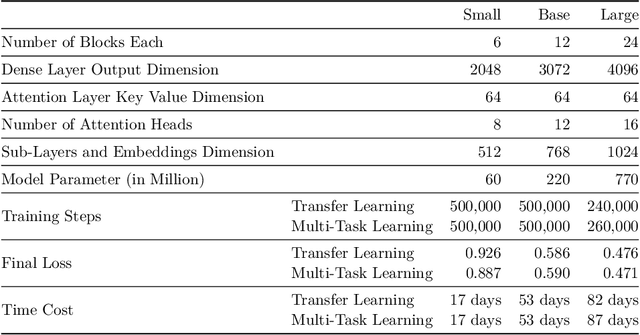
Currently, a growing number of mature natural language processing applications make people's life more convenient. Such applications are built by source code - the language in software engineering. However, the applications for understanding source code language to ease the software engineering process are under-researched. Simultaneously, the transformer model, especially its combination with transfer learning, has been proven to be a powerful technique for natural language processing tasks. These breakthroughs point out a promising direction for process source code and crack software engineering tasks. This paper describes CodeTrans - an encoder-decoder transformer model for tasks in the software engineering domain, that explores the effectiveness of encoder-decoder transformer models for six software engineering tasks, including thirteen sub-tasks. Moreover, we have investigated the effect of different training strategies, including single-task learning, transfer learning, multi-task learning, and multi-task learning with fine-tuning. CodeTrans outperforms the state-of-the-art models on all the tasks. To expedite future works in the software engineering domain, we have published our pre-trained models of CodeTrans. https://github.com/agemagician/CodeTrans
ProtTrans: Towards Cracking the Language of Life's Code Through Self-Supervised Deep Learning and High Performance Computing
Jul 20, 2020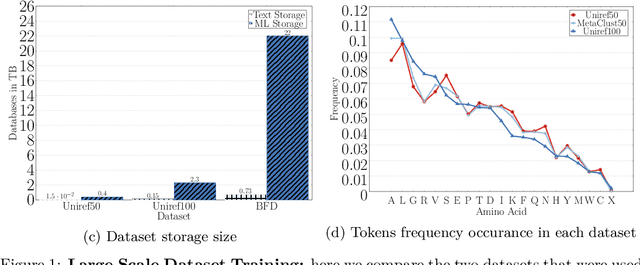
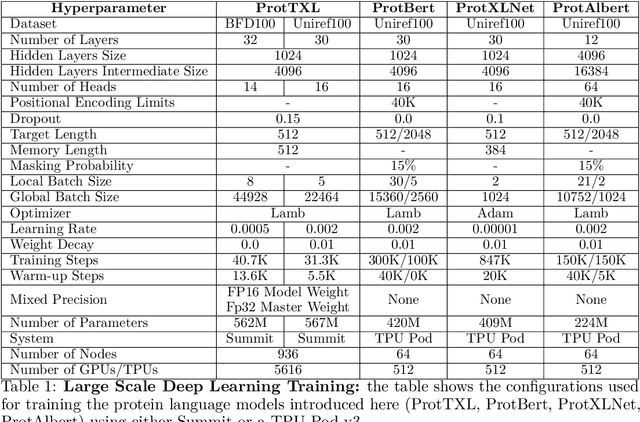
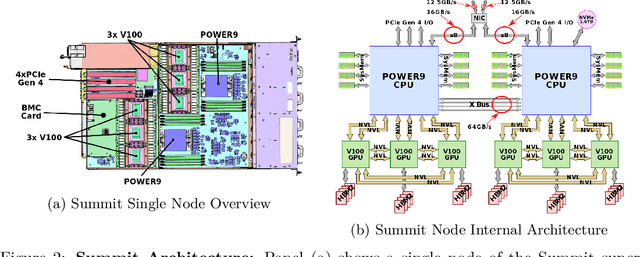
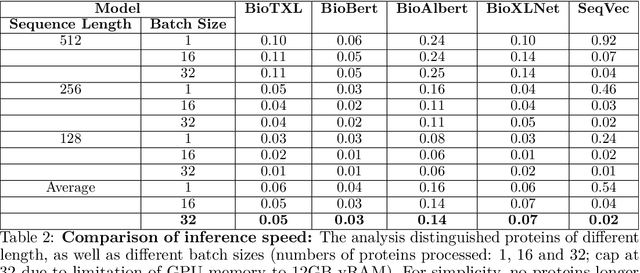
Computational biology and bioinformatics provide vast data gold-mines from protein sequences, ideal for Language Models (LMs) taken from Natural Language Processing (NLP). These LMs reach for new prediction frontiers at low inference costs. Here, we trained two auto-regressive language models (Transformer-XL, XLNet) and two auto-encoder models (Bert, Albert) on data from UniRef and BFD containing up to 393 billion amino acids (words) from 2.1 billion protein sequences (22- and 112-times the entire English Wikipedia). The LMs were trained on the Summit supercomputer at Oak Ridge National Laboratory (ORNL), using 936 nodes (total 5616 GPUs) and one TPU Pod (V3-512 or V3-1024). We validated the advantage of up-scaling LMs to larger models supported by bigger data by predicting secondary structure (3-states: Q3=76-84, 8-states: Q8=65-73), sub-cellular localization for 10 cellular compartments (Q10=74) and whether a protein is membrane-bound or water-soluble (Q2=89). Dimensionality reduction revealed that the LM-embeddings from unlabeled data (only protein sequences) captured important biophysical properties governing protein shape. This implied learning some of the grammar of the language of life realized in protein sequences. The successful up-scaling of protein LMs through HPC to larger data sets slightly reduced the gap between models trained on evolutionary information and LMs. The official GitHub repository: https://github.com/agemagician/ProtTrans