 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRePanda: Pandas-powered Tabular Verification and Reasoning
Mar 14, 2025Fact-checking tabular data is essential for ensuring the accuracy of structured information. However, existing methods often rely on black-box models with opaque reasoning. We introduce RePanda, a structured fact verification approach that translates claims into executable pandas queries, enabling interpretable and verifiable reasoning. To train RePanda, we construct PanTabFact, a structured dataset derived from the TabFact train set, where claims are paired with executable queries generated using DeepSeek-Chat and refined through automated error correction. Fine-tuning DeepSeek-coder-7B-instruct-v1.5 on PanTabFact, RePanda achieves 84.09% accuracy on the TabFact test set. To evaluate Out-of-Distribution (OOD) generalization, we interpret question-answer pairs from WikiTableQuestions as factual claims and refer to this dataset as WikiFact. Without additional fine-tuning, RePanda achieves 84.72% accuracy on WikiFact, significantly outperforming all other baselines and demonstrating strong OOD robustness. Notably, these results closely match the zero-shot performance of DeepSeek-Chat (671B), indicating that our fine-tuning approach effectively distills structured reasoning from a much larger model into a compact, locally executable 7B model. Beyond fact verification, RePanda extends to tabular question answering by generating executable queries that retrieve precise answers. To support this, we introduce PanWiki, a dataset mapping WikiTableQuestions to pandas queries. Fine-tuning on PanWiki, RePanda achieves 75.1% accuracy in direct answer retrieval. These results highlight the effectiveness of structured execution-based reasoning for tabular verification and question answering. We have publicly released the dataset on Hugging Face at datasets/AtoosaChegini/PanTabFact.
EditVal: Benchmarking Diffusion Based Text-Guided Image Editing Methods
Oct 03, 2023
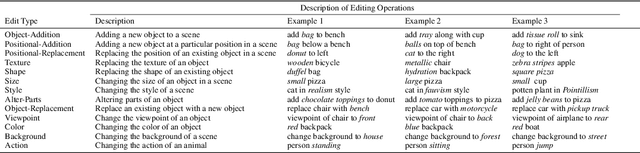
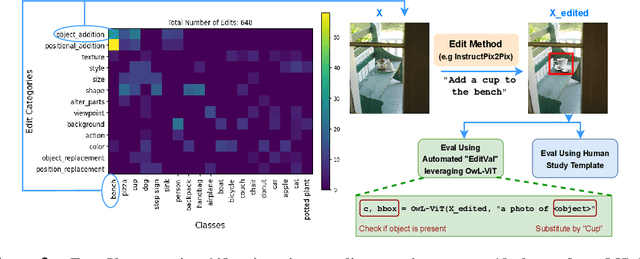
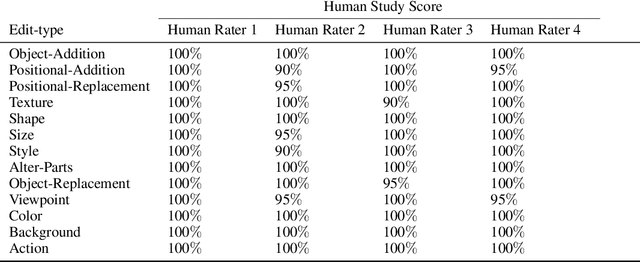
A plethora of text-guided image editing methods have recently been developed by leveraging the impressive capabilities of large-scale diffusion-based generative models such as Imagen and Stable Diffusion. A standardized evaluation protocol, however, does not exist to compare methods across different types of fine-grained edits. To address this gap, we introduce EditVal, a standardized benchmark for quantitatively evaluating text-guided image editing methods. EditVal consists of a curated dataset of images, a set of editable attributes for each image drawn from 13 possible edit types, and an automated evaluation pipeline that uses pre-trained vision-language models to assess the fidelity of generated images for each edit type. We use EditVal to benchmark 8 cutting-edge diffusion-based editing methods including SINE, Imagic and Instruct-Pix2Pix. We complement this with a large-scale human study where we show that EditVall's automated evaluation pipeline is strongly correlated with human-preferences for the edit types we considered. From both the human study and automated evaluation, we find that: (i) Instruct-Pix2Pix, Null-Text and SINE are the top-performing methods averaged across different edit types, however {\it only} Instruct-Pix2Pix and Null-Text are able to preserve original image properties; (ii) Most of the editing methods fail at edits involving spatial operations (e.g., changing the position of an object). (iii) There is no `winner' method which ranks the best individually across a range of different edit types. We hope that our benchmark can pave the way to developing more reliable text-guided image editing tools in the future. We will publicly release EditVal, and all associated code and human-study templates to support these research directions in https://deep-ml-research.github.io/editval/.
Data-Centric Debugging: mitigating model failures via targeted data collection
Nov 17, 2022



Deep neural networks can be unreliable in the real world when the training set does not adequately cover all the settings where they are deployed. Focusing on image classification, we consider the setting where we have an error distribution $\mathcal{E}$ representing a deployment scenario where the model fails. We have access to a small set of samples $\mathcal{E}_{sample}$ from $\mathcal{E}$ and it can be expensive to obtain additional samples. In the traditional model development framework, mitigating failures of the model in $\mathcal{E}$ can be challenging and is often done in an ad hoc manner. In this paper, we propose a general methodology for model debugging that can systemically improve model performance on $\mathcal{E}$ while maintaining its performance on the original test set. Our key assumption is that we have access to a large pool of weakly (noisily) labeled data $\mathcal{F}$. However, naively adding $\mathcal{F}$ to the training would hurt model performance due to the large extent of label noise. Our Data-Centric Debugging (DCD) framework carefully creates a debug-train set by selecting images from $\mathcal{F}$ that are perceptually similar to the images in $\mathcal{E}_{sample}$. To do this, we use the $\ell_2$ distance in the feature space (penultimate layer activations) of various models including ResNet, Robust ResNet and DINO where we observe DINO ViTs are significantly better at discovering similar images compared to Resnets. Compared to LPIPS, we find that our method reduces compute and storage requirements by 99.58\%. Compared to the baselines that maintain model performance on the test set, we achieve significantly (+9.45\%) improved results on the debug-heldout sets.
InForecaster: Forecasting Influenza Hemagglutinin Mutations Through the Lens of Anomaly Detection
Oct 25, 2022The influenza virus hemagglutinin is an important part of the virus attachment to the host cells. The hemagglutinin proteins are one of the genetic regions of the virus with a high potential for mutations. Due to the importance of predicting mutations in producing effective and low-cost vaccines, solutions that attempt to approach this problem have recently gained a significant attention. A historical record of mutations have been used to train predictive models in such solutions. However, the imbalance between mutations and the preserved proteins is a big challenge for the development of such models that needs to be addressed. Here, we propose to tackle this challenge through anomaly detection (AD). AD is a well-established field in Machine Learning (ML) that tries to distinguish unseen anomalies from the normal patterns using only normal training samples. By considering mutations as the anomalous behavior, we could benefit existing rich solutions in this field that have emerged recently. Such methods also fit the problem setup of extreme imbalance between the number of unmutated vs. mutated training samples. Motivated by this formulation, our method tries to find a compact representation for unmutated samples while forcing anomalies to be separated from the normal ones. This helps the model to learn a shared unique representation between normal training samples as much as possible, which improves the discernibility and detectability of mutated samples from the unmutated ones at the test time. We conduct a large number of experiments on four publicly available datasets, consisting of 3 different hemagglutinin protein datasets, and one SARS-CoV-2 dataset, and show the effectiveness of our method through different standard criteria.