 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChemPile: A 250GB Diverse and Curated Dataset for Chemical Foundation Models
May 18, 2025



Foundation models have shown remarkable success across scientific domains, yet their impact in chemistry remains limited due to the absence of diverse, large-scale, high-quality datasets that reflect the field's multifaceted nature. We present the ChemPile, an open dataset containing over 75 billion tokens of curated chemical data, specifically built for training and evaluating general-purpose models in the chemical sciences. The dataset mirrors the human learning journey through chemistry -- from educational foundations to specialized expertise -- spanning multiple modalities and content types including structured data in diverse chemical representations (SMILES, SELFIES, IUPAC names, InChI, molecular renderings), scientific and educational text, executable code, and chemical images. ChemPile integrates foundational knowledge (textbooks, lecture notes), specialized expertise (scientific articles and language-interfaced data), visual understanding (molecular structures, diagrams), and advanced reasoning (problem-solving traces and code) -- mirroring how human chemists develop expertise through diverse learning materials and experiences. Constructed through hundreds of hours of expert curation, the ChemPile captures both foundational concepts and domain-specific complexity. We provide standardized training, validation, and test splits, enabling robust benchmarking. ChemPile is openly released via HuggingFace with a consistent API, permissive license, and detailed documentation. We hope the ChemPile will serve as a catalyst for chemical AI, enabling the development of the next generation of chemical foundation models.
MassSpecGym: A benchmark for the discovery and identification of molecules
Oct 30, 2024The discovery and identification of molecules in biological and environmental samples is crucial for advancing biomedical and chemical sciences. Tandem mass spectrometry (MS/MS) is the leading technique for high-throughput elucidation of molecular structures. However, decoding a molecular structure from its mass spectrum is exceptionally challenging, even when performed by human experts. As a result, the vast majority of acquired MS/MS spectra remain uninterpreted, thereby limiting our understanding of the underlying (bio)chemical processes. Despite decades of progress in machine learning applications for predicting molecular structures from MS/MS spectra, the development of new methods is severely hindered by the lack of standard datasets and evaluation protocols. To address this problem, we propose MassSpecGym -- the first comprehensive benchmark for the discovery and identification of molecules from MS/MS data. Our benchmark comprises the largest publicly available collection of high-quality labeled MS/MS spectra and defines three MS/MS annotation challenges: \textit{de novo} molecular structure generation, molecule retrieval, and spectrum simulation. It includes new evaluation metrics and a generalization-demanding data split, therefore standardizing the MS/MS annotation tasks and rendering the problem accessible to the broad machine learning community. MassSpecGym is publicly available at \url{https://github.com/pluskal-lab/MassSpecGym}.
FraGNNet: A Deep Probabilistic Model for Mass Spectrum Prediction
Apr 02, 2024



The process of identifying a compound from its mass spectrum is a critical step in the analysis of complex mixtures. Typical solutions for the mass spectrum to compound (MS2C) problem involve matching the unknown spectrum against a library of known spectrum-molecule pairs, an approach that is limited by incomplete library coverage. Compound to mass spectrum (C2MS) models can improve retrieval rates by augmenting real libraries with predicted spectra. Unfortunately, many existing C2MS models suffer from problems with prediction resolution, scalability, or interpretability. We develop a new probabilistic method for C2MS prediction, FraGNNet, that can efficiently and accurately predict high-resolution spectra. FraGNNet uses a structured latent space to provide insight into the underlying processes that define the spectrum. Our model achieves state-of-the-art performance in terms of prediction error, and surpasses existing C2MS models as a tool for retrieval-based MS2C.
Unleashing the Strengths of Unlabeled Data in Pan-cancer Abdominal Organ Quantification: the FLARE22 Challenge
Aug 10, 2023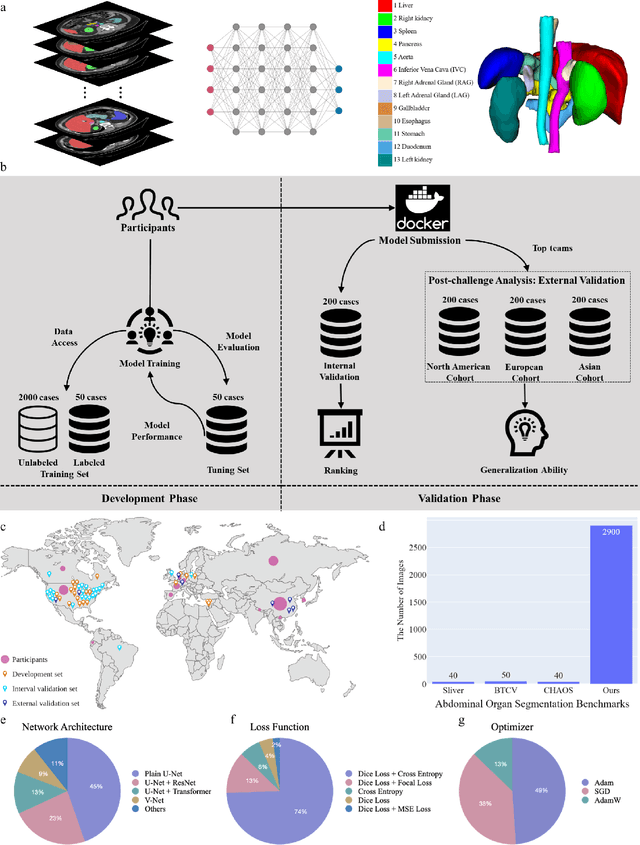
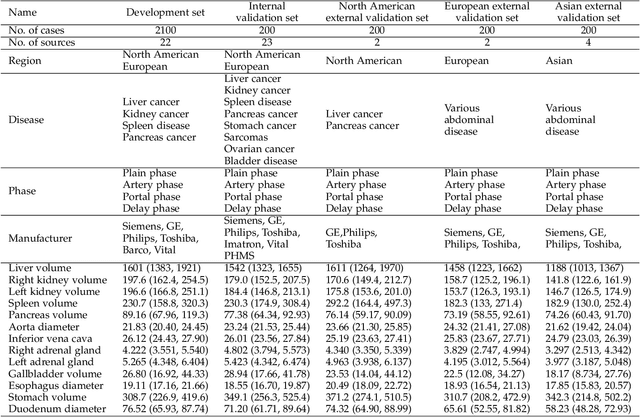
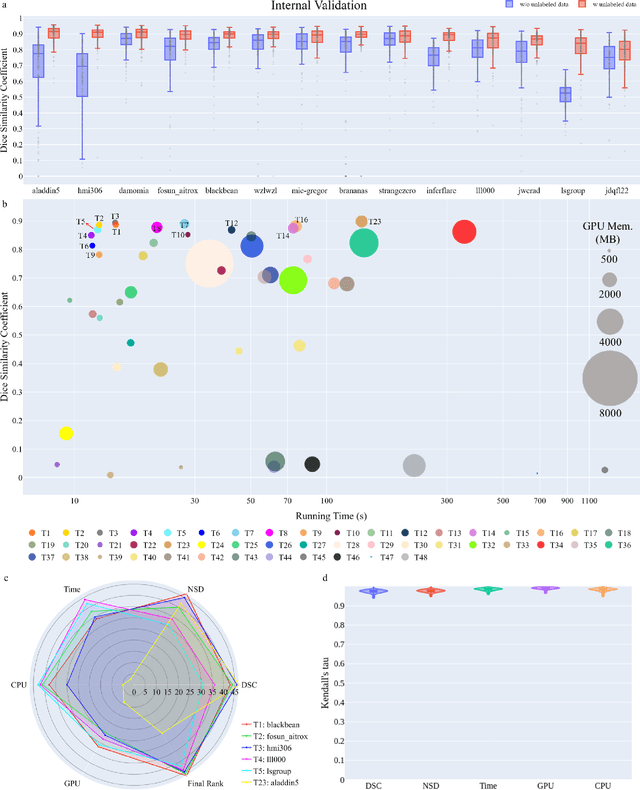
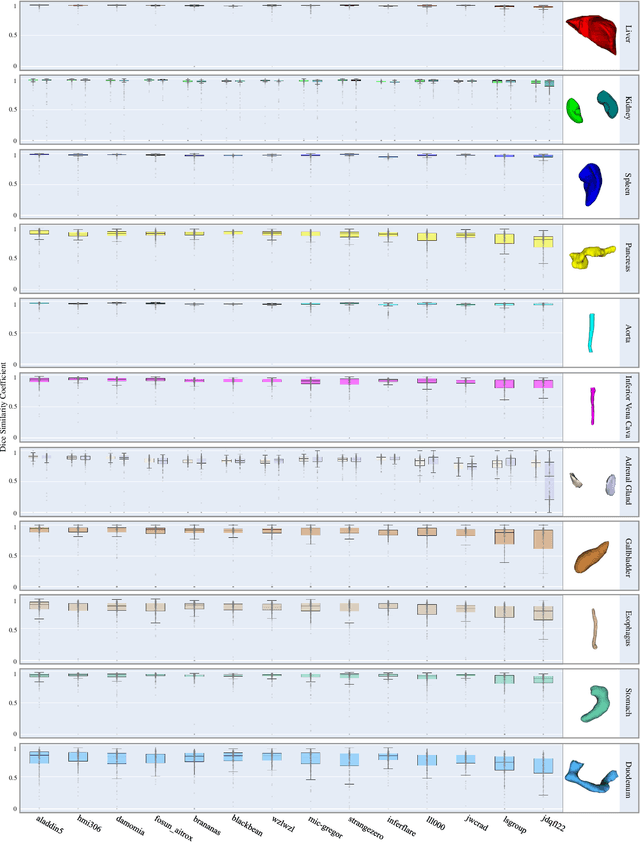
Quantitative organ assessment is an essential step in automated abdominal disease diagnosis and treatment planning. Artificial intelligence (AI) has shown great potential to automatize this process. However, most existing AI algorithms rely on many expert annotations and lack a comprehensive evaluation of accuracy and efficiency in real-world multinational settings. To overcome these limitations, we organized the FLARE 2022 Challenge, the largest abdominal organ analysis challenge to date, to benchmark fast, low-resource, accurate, annotation-efficient, and generalized AI algorithms. We constructed an intercontinental and multinational dataset from more than 50 medical groups, including Computed Tomography (CT) scans with different races, diseases, phases, and manufacturers. We independently validated that a set of AI algorithms achieved a median Dice Similarity Coefficient (DSC) of 90.0\% by using 50 labeled scans and 2000 unlabeled scans, which can significantly reduce annotation requirements. The best-performing algorithms successfully generalized to holdout external validation sets, achieving a median DSC of 89.5\%, 90.9\%, and 88.3\% on North American, European, and Asian cohorts, respectively. They also enabled automatic extraction of key organ biology features, which was labor-intensive with traditional manual measurements. This opens the potential to use unlabeled data to boost performance and alleviate annotation shortages for modern AI models.
SELFIES and the future of molecular string representations
Mar 31, 2022
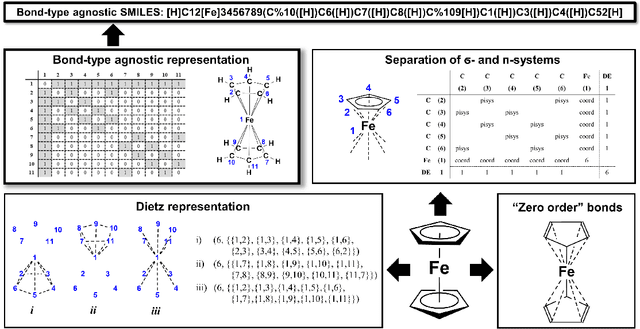
Artificial intelligence (AI) and machine learning (ML) are expanding in popularity for broad applications to challenging tasks in chemistry and materials science. Examples include the prediction of properties, the discovery of new reaction pathways, or the design of new molecules. The machine needs to read and write fluently in a chemical language for each of these tasks. Strings are a common tool to represent molecular graphs, and the most popular molecular string representation, SMILES, has powered cheminformatics since the late 1980s. However, in the context of AI and ML in chemistry, SMILES has several shortcomings -- most pertinently, most combinations of symbols lead to invalid results with no valid chemical interpretation. To overcome this issue, a new language for molecules was introduced in 2020 that guarantees 100\% robustness: SELFIES (SELF-referencIng Embedded Strings). SELFIES has since simplified and enabled numerous new applications in chemistry. In this manuscript, we look to the future and discuss molecular string representations, along with their respective opportunities and challenges. We propose 16 concrete Future Projects for robust molecular representations. These involve the extension toward new chemical domains, exciting questions at the interface of AI and robust languages and interpretability for both humans and machines. We hope that these proposals will inspire several follow-up works exploiting the full potential of molecular string representations for the future of AI in chemistry and materials science.
MassFormer: Tandem Mass Spectrum Prediction with Graph Transformers
Nov 15, 2021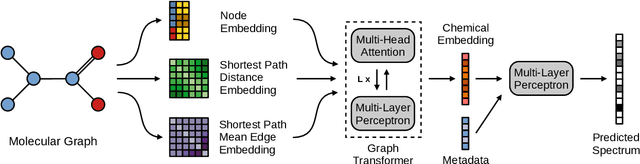
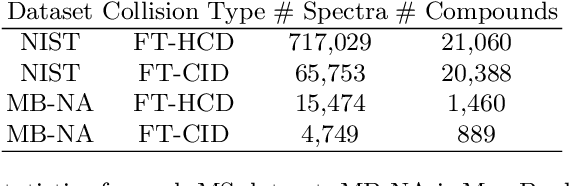
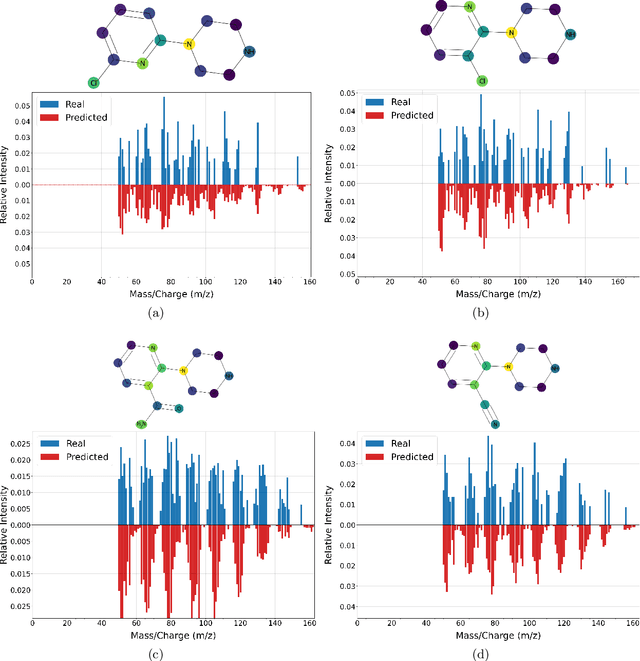
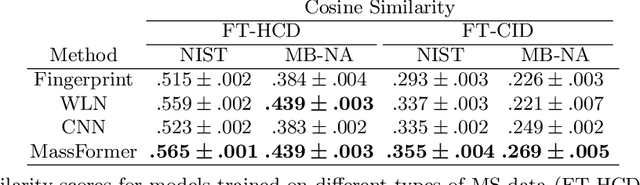
Mass spectrometry is a key tool in the study of small molecules, playing an important role in metabolomics, drug discovery, and environmental chemistry. Tandem mass spectra capture fragmentation patterns that provide key structural information about a molecule and help with its identification. Practitioners often rely on spectral library searches to match unknown spectra with known compounds. However, such search-based methods are limited by availability of reference experimental data. In this work we show that graph transformers can be used to accurately predict tandem mass spectra. Our model, MassFormer, outperforms competing deep learning approaches for spectrum prediction, and includes an interpretable attention mechanism to help explain predictions. We demonstrate that our model can be used to improve reference library coverage on a synthetic molecule identification task. Through quantitative analysis and visual inspection, we verify that our model recovers prior knowledge about the effect of collision energy on the generated spectrum. We evaluate our model on different types of mass spectra from two independent MS datasets and show that its performance generalizes. Code available at github.com/Roestlab/massformer.