 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTraining on test proteins improves fitness, structure, and function prediction
Nov 04, 2024



Data scarcity and distribution shifts often hinder the ability of machine learning models to generalize when applied to proteins and other biological data. Self-supervised pre-training on large datasets is a common method to enhance generalization. However, striving to perform well on all possible proteins can limit model's capacity to excel on any specific one, even though practitioners are often most interested in accurate predictions for the individual protein they study. To address this limitation, we propose an orthogonal approach to achieve generalization. Building on the prevalence of self-supervised pre-training, we introduce a method for self-supervised fine-tuning at test time, allowing models to adapt to the test protein of interest on the fly and without requiring any additional data. We study our test-time training (TTT) method through the lens of perplexity minimization and show that it consistently enhances generalization across different models, their scales, and datasets. Notably, our method leads to new state-of-the-art results on the standard benchmark for protein fitness prediction, improves protein structure prediction for challenging targets, and enhances function prediction accuracy.
MassSpecGym: A benchmark for the discovery and identification of molecules
Oct 30, 2024The discovery and identification of molecules in biological and environmental samples is crucial for advancing biomedical and chemical sciences. Tandem mass spectrometry (MS/MS) is the leading technique for high-throughput elucidation of molecular structures. However, decoding a molecular structure from its mass spectrum is exceptionally challenging, even when performed by human experts. As a result, the vast majority of acquired MS/MS spectra remain uninterpreted, thereby limiting our understanding of the underlying (bio)chemical processes. Despite decades of progress in machine learning applications for predicting molecular structures from MS/MS spectra, the development of new methods is severely hindered by the lack of standard datasets and evaluation protocols. To address this problem, we propose MassSpecGym -- the first comprehensive benchmark for the discovery and identification of molecules from MS/MS data. Our benchmark comprises the largest publicly available collection of high-quality labeled MS/MS spectra and defines three MS/MS annotation challenges: \textit{de novo} molecular structure generation, molecule retrieval, and spectrum simulation. It includes new evaluation metrics and a generalization-demanding data split, therefore standardizing the MS/MS annotation tasks and rendering the problem accessible to the broad machine learning community. MassSpecGym is publicly available at \url{https://github.com/pluskal-lab/MassSpecGym}.
Revealing data leakage in protein interaction benchmarks
Apr 16, 2024In recent years, there has been remarkable progress in machine learning for protein-protein interactions. However, prior work has predominantly focused on improving learning algorithms, with less attention paid to evaluation strategies and data preparation. Here, we demonstrate that further development of machine learning methods may be hindered by the quality of existing train-test splits. Specifically, we find that commonly used splitting strategies for protein complexes, based on protein sequence or metadata similarity, introduce major data leakage. This may result in overoptimistic evaluation of generalization, as well as unfair benchmarking of the models, biased towards assessing their overfitting capacity rather than practical utility. To overcome the data leakage, we recommend constructing data splits based on 3D structural similarity of protein-protein interfaces and suggest corresponding algorithms. We believe that addressing the data leakage problem is critical for further progress in this research area.
Learning to design protein-protein interactions with enhanced generalization
Oct 27, 2023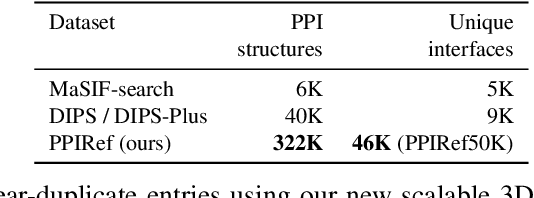
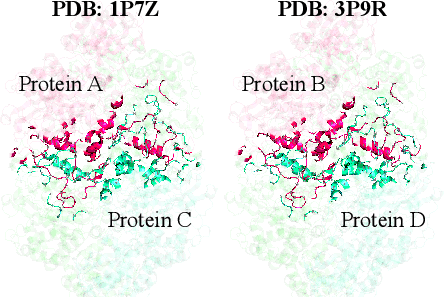
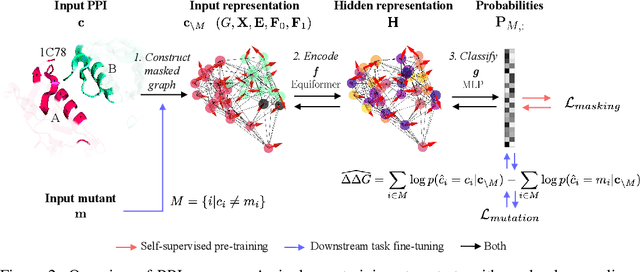
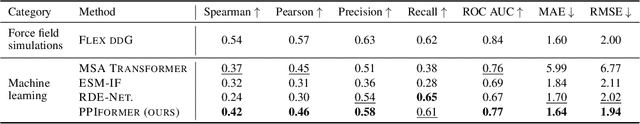
Discovering mutations enhancing protein-protein interactions (PPIs) is critical for advancing biomedical research and developing improved therapeutics. While machine learning approaches have substantially advanced the field, they often struggle to generalize beyond training data in practical scenarios. The contributions of this work are three-fold. First, we construct PPIRef, the largest and non-redundant dataset of 3D protein-protein interactions, enabling effective large-scale learning. Second, we leverage the PPIRef dataset to pre-train PPIformer, a new SE(3)-equivariant model generalizing across diverse protein-binder variants. We fine-tune PPIformer to predict effects of mutations on protein-protein interactions via a thermodynamically motivated adjustment of the pre-training loss function. Finally, we demonstrate the enhanced generalization of our new PPIformer approach by outperforming other state-of-the-art methods on new, non-leaking splits of standard labeled PPI mutational data and independent case studies optimizing a human antibody against SARS-CoV-2 and increasing the thrombolytic activity of staphylokinase.