 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMSDNet: Efficient 4D Radar Super-Resolution via Multi-Stage Distillation
Sep 16, 2025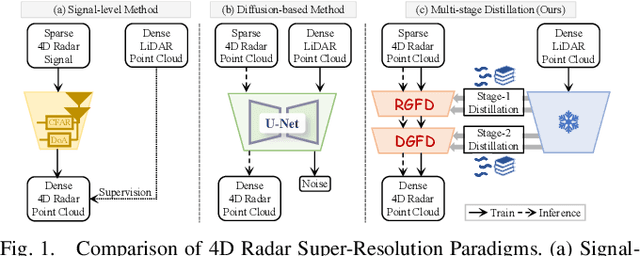
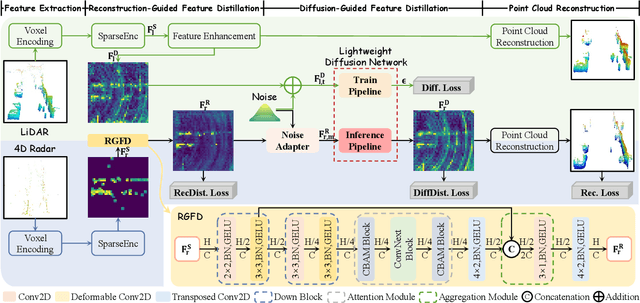
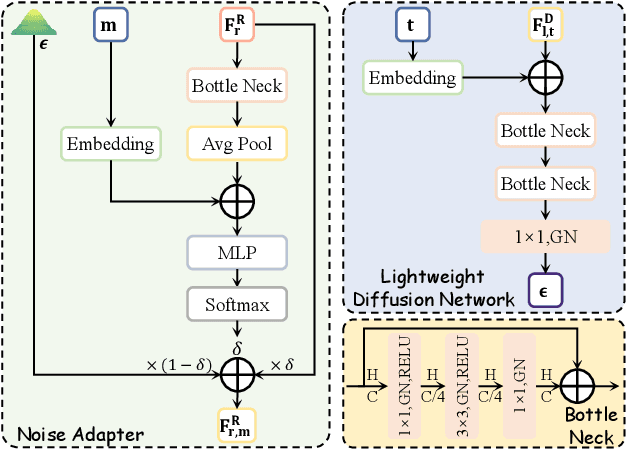
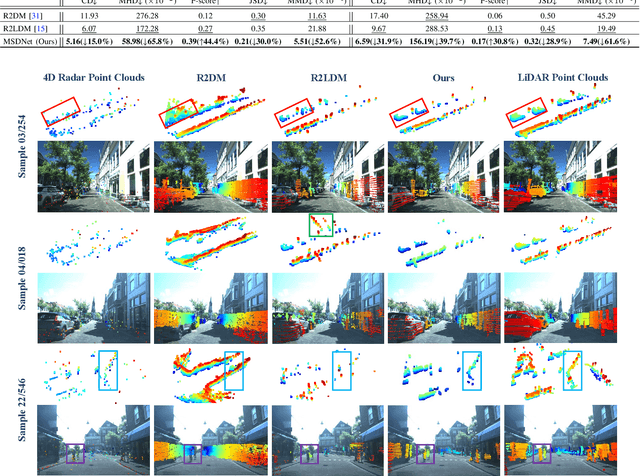
4D radar super-resolution, which aims to reconstruct sparse and noisy point clouds into dense and geometrically consistent representations, is a foundational problem in autonomous perception. However, existing methods often suffer from high training cost or rely on complex diffusion-based sampling, resulting in high inference latency and poor generalization, making it difficult to balance accuracy and efficiency. To address these limitations, we propose MSDNet, a multi-stage distillation framework that efficiently transfers dense LiDAR priors to 4D radar features to achieve both high reconstruction quality and computational efficiency. The first stage performs reconstruction-guided feature distillation, aligning and densifying the student's features through feature reconstruction. In the second stage, we propose diffusion-guided feature distillation, which treats the stage-one distilled features as a noisy version of the teacher's representations and refines them via a lightweight diffusion network. Furthermore, we introduce a noise adapter that adaptively aligns the noise level of the feature with a predefined diffusion timestep, enabling a more precise denoising. Extensive experiments on the VoD and in-house datasets demonstrate that MSDNet achieves both high-fidelity reconstruction and low-latency inference in the task of 4D radar point cloud super-resolution, and consistently improves performance on downstream tasks. The code will be publicly available upon publication.
Forecasting Unseen Points of Interest Visits Using Context and Proximity Priors
Nov 22, 2024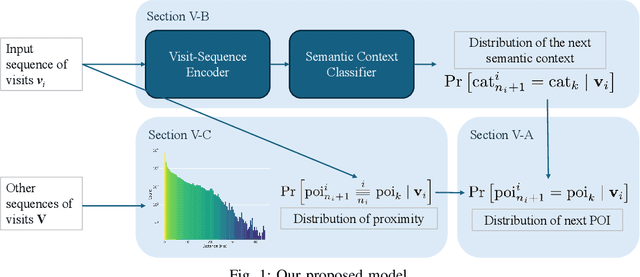
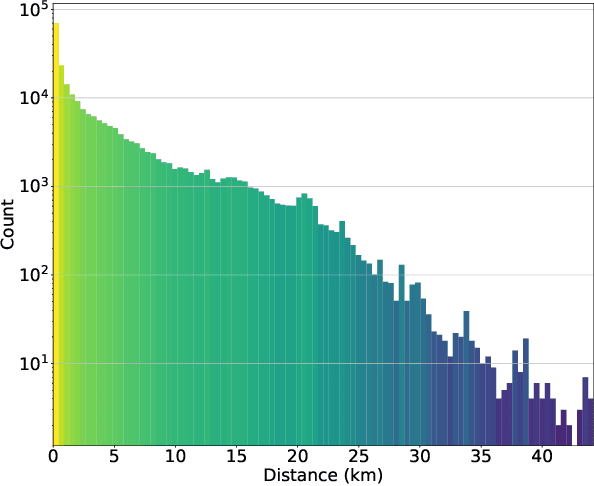
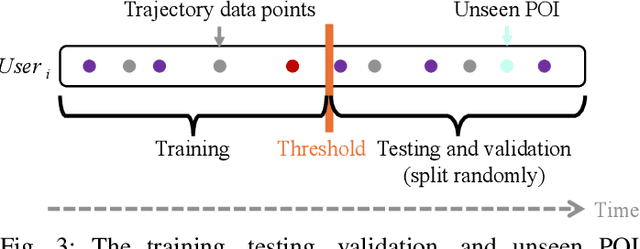
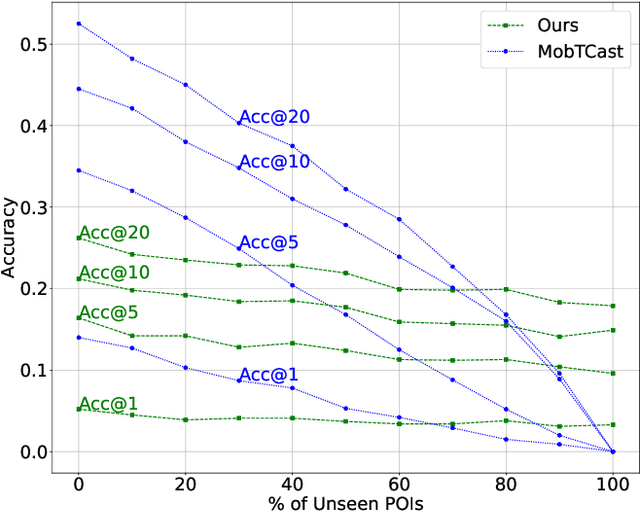
Understanding human mobility behavior is crucial for numerous applications, including crowd management, location-based recommendations, and the estimation of pandemic spread. Machine learning models can predict the Points of Interest (POIs) that individuals are likely to visit in the future by analyzing their historical visit patterns. Previous studies address this problem by learning a POI classifier, where each class corresponds to a POI. However, this limits their applicability to predict a new POI that was not in the training data, such as the opening of new restaurants. To address this challenge, we propose a model designed to predict a new POI outside the training data as long as its context is aligned with the user's interests. Unlike existing approaches that directly predict specific POIs, our model first forecasts the semantic context of potential future POIs, then combines this with a proximity-based prior probability distribution to determine the exact POI. Experimental results on real-world visit data demonstrate that our model outperforms baseline methods that do not account for semantic contexts, achieving a 17% improvement in accuracy. Notably, as new POIs are introduced over time, our model remains robust, exhibiting a lower decline rate in prediction accuracy compared to existing methods.
Kolmogorov-Arnold Networks are Radial Basis Function Networks
May 10, 2024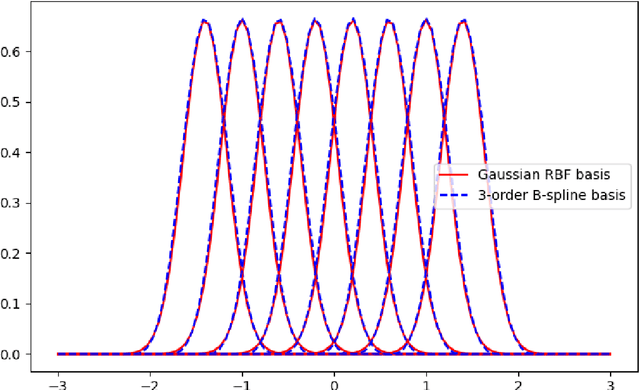
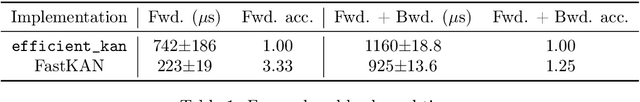
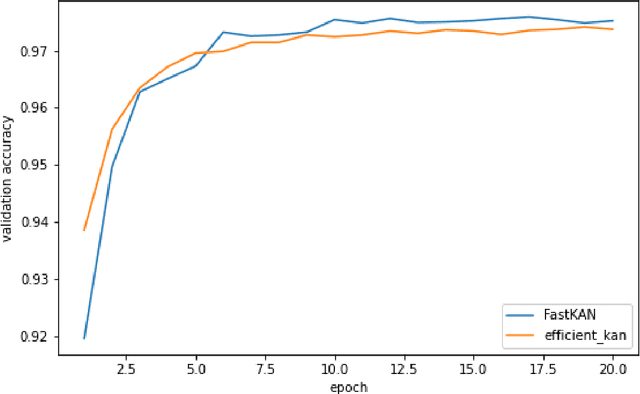
This short paper is a fast proof-of-concept that the 3-order B-splines used in Kolmogorov-Arnold Networks (KANs) can be well approximated by Gaussian radial basis functions. Doing so leads to FastKAN, a much faster implementation of KAN which is also a radial basis function (RBF) network.
Deep Molecular Representation Learning via Fusing Physical and Chemical Information
Nov 28, 2021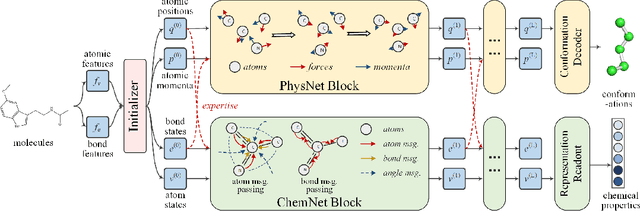
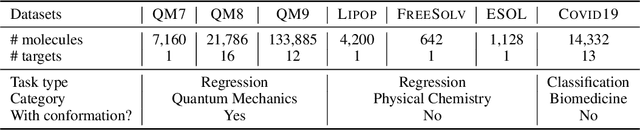
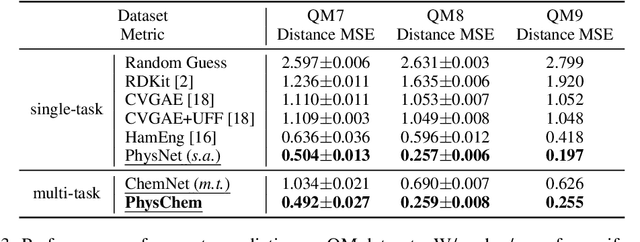
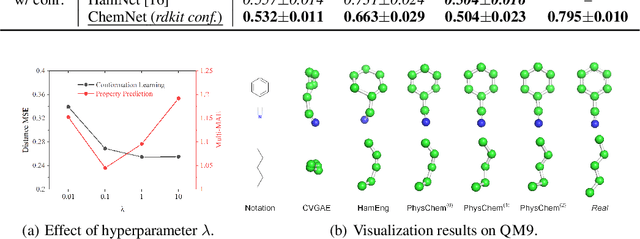
Molecular representation learning is the first yet vital step in combining deep learning and molecular science. To push the boundaries of molecular representation learning, we present PhysChem, a novel neural architecture that learns molecular representations via fusing physical and chemical information of molecules. PhysChem is composed of a physicist network (PhysNet) and a chemist network (ChemNet). PhysNet is a neural physical engine that learns molecular conformations through simulating molecular dynamics with parameterized forces; ChemNet implements geometry-aware deep message-passing to learn chemical / biomedical properties of molecules. Two networks specialize in their own tasks and cooperate by providing expertise to each other. By fusing physical and chemical information, PhysChem achieved state-of-the-art performances on MoleculeNet, a standard molecular machine learning benchmark. The effectiveness of PhysChem was further corroborated on cutting-edge datasets of SARS-CoV-2.
HamNet: Conformation-Guided Molecular Representation with Hamiltonian Neural Networks
May 08, 2021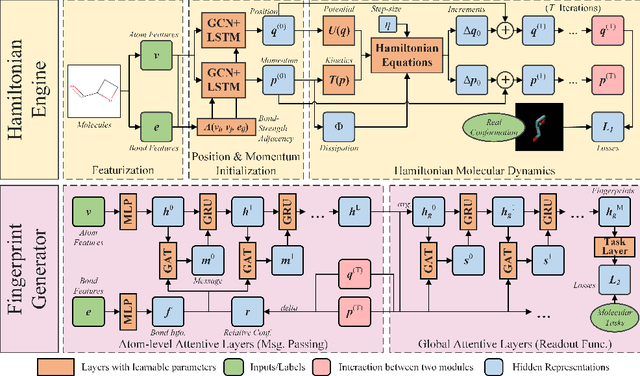
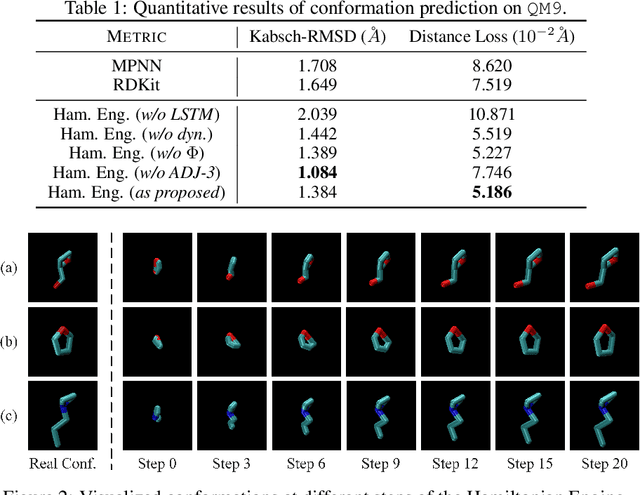
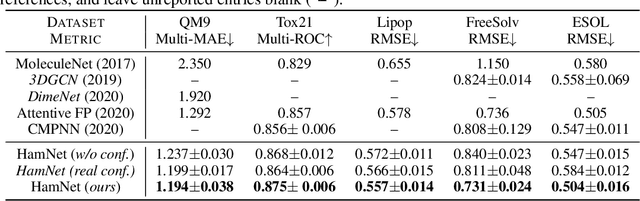
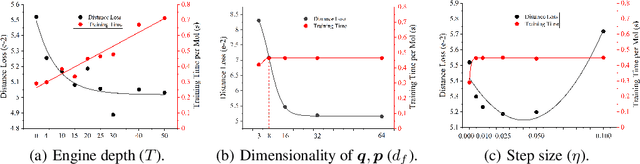
Well-designed molecular representations (fingerprints) are vital to combine medical chemistry and deep learning. Whereas incorporating 3D geometry of molecules (i.e. conformations) in their representations seems beneficial, current 3D algorithms are still in infancy. In this paper, we propose a novel molecular representation algorithm which preserves 3D conformations of molecules with a Molecular Hamiltonian Network (HamNet). In HamNet, implicit positions and momentums of atoms in a molecule interact in the Hamiltonian Engine following the discretized Hamiltonian equations. These implicit coordinations are supervised with real conformations with translation- & rotation-invariant losses, and further used as inputs to the Fingerprint Generator, a message-passing neural network. Experiments show that the Hamiltonian Engine can well preserve molecular conformations, and that the fingerprints generated by HamNet achieve state-of-the-art performances on MoleculeNet, a standard molecular machine learning benchmark.
Learning Node Representations from Noisy Graph Structures
Dec 04, 2020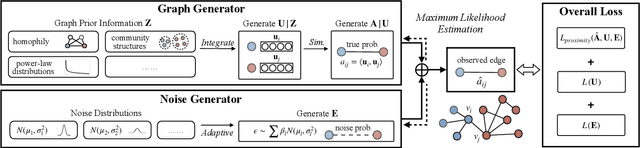
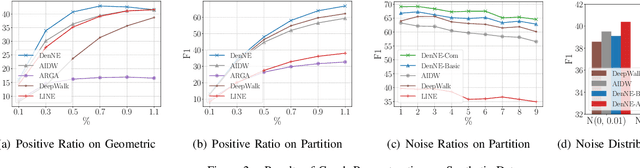
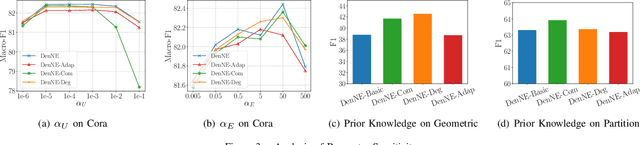
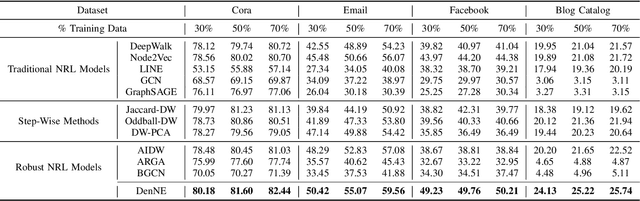
Learning low-dimensional representations on graphs has proved to be effective in various downstream tasks. However, noises prevail in real-world networks, which compromise networks to a large extent in that edges in networks propagate noises through the whole network instead of only the node itself. While existing methods tend to focus on preserving structural properties, the robustness of the learned representations against noises is generally ignored. In this paper, we propose a novel framework to learn noise-free node representations and eliminate noises simultaneously. Since noises are often unknown on real graphs, we design two generators, namely a graph generator and a noise generator, to identify normal structures and noises in an unsupervised setting. On the one hand, the graph generator serves as a unified scheme to incorporate any useful graph prior knowledge to generate normal structures. We illustrate the generative process with community structures and power-law degree distributions as examples. On the other hand, the noise generator generates graph noises not only satisfying some fundamental properties but also in an adaptive way. Thus, real noises with arbitrary distributions can be handled successfully. Finally, in order to eliminate noises and obtain noise-free node representations, two generators need to be optimized jointly, and through maximum likelihood estimation, we equivalently convert the model into imposing different regularization constraints on the true graph and noises respectively. Our model is evaluated on both real-world and synthetic data. It outperforms other strong baselines for node classification and graph reconstruction tasks, demonstrating its ability to eliminate graph noises.
A Novel User Representation Paradigm for Making Personalized Candidate Retrieval
Jul 15, 2019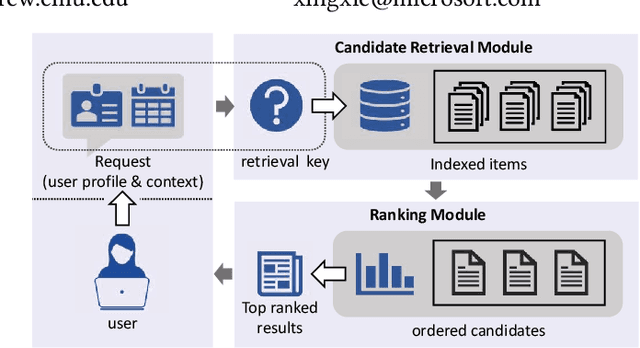
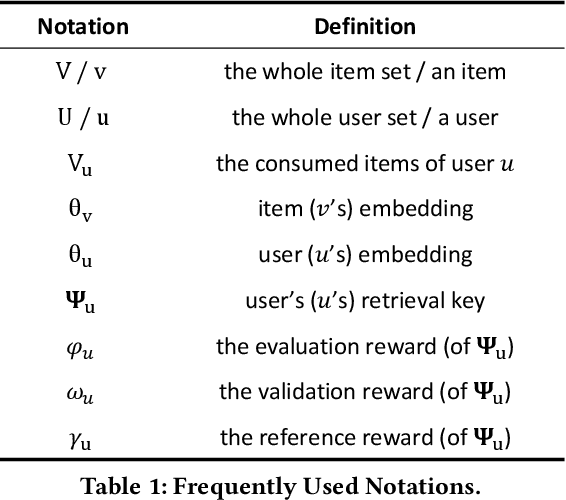
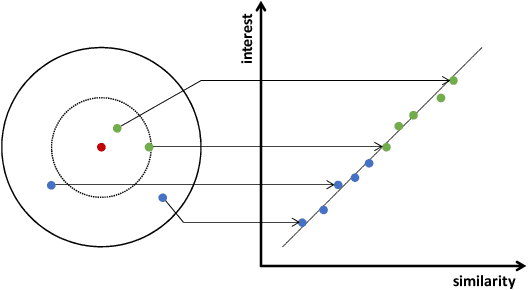
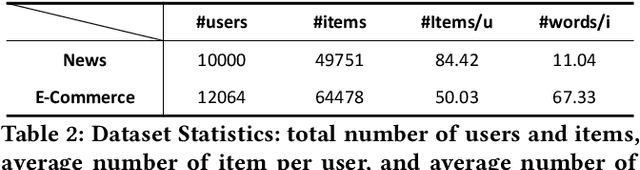
Candidate retrieval is a crucial part in recommendation system, where quality candidates need to be selected in realtime for user's recommendation request. Conventional methods would make use of feature similarity directly for highly scalable retrieval, yet their retrieval quality can be limited due to inferior user interest modeling. In contrast, deep learning-based recommenders are precise in modeling user interest, but they are difficult to be scaled for efficient candidate retrieval. In this work, a novel paradigm Synthonet is proposed for both precise and scalable candidate retrieval. With Synthonet, user is represented as a compact vector known as retrieval key. By developing an Actor-Critic learning framework, the generation of retrieval key is optimally conducted, such that the similarity between retrieval key and item's representation will accurately reflect user's interest towards the corresponding item. Consequently, quality candidates can be acquired in realtime on top of highly efficient similarity search methods. Comprehensive empirical studies are carried out for the verification of our proposed methods, where consistent and remarkable improvements are achieved over a series of competitive baselines, including representative variations on metric learning.
GCN-LASE: Towards Adequately Incorporating Link Attributes in Graph Convolutional Networks
Feb 26, 2019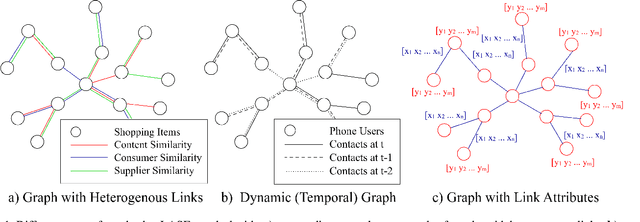
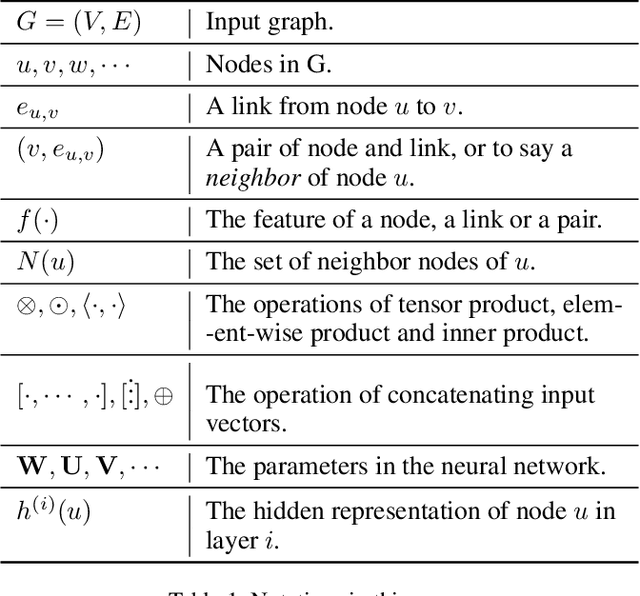
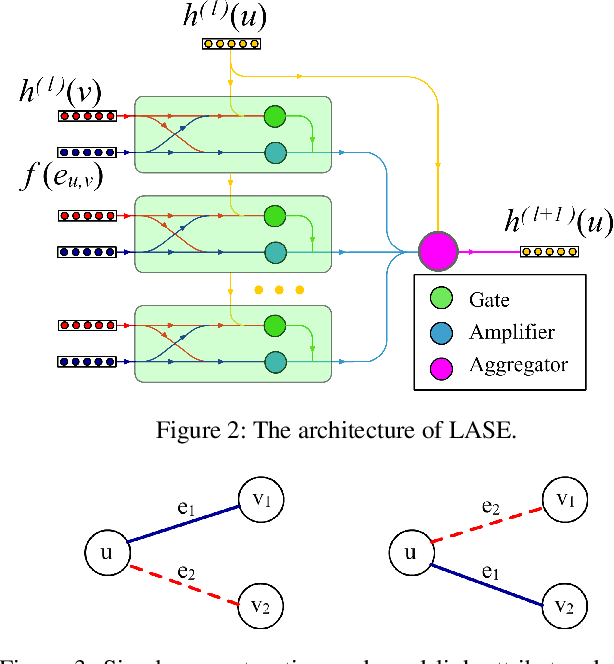
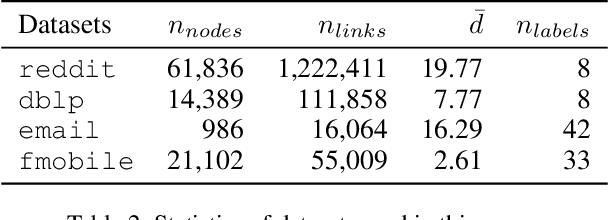
Graph Convolutional Networks (GCNs) have proved to be a most powerful architecture in aggregating local neighborhood information for individual graph nodes. Low-rank proximities and node features are successfully leveraged in existing GCNs, however, attributes that graph links may carry are commonly ignored, as almost all of these models simplify graph links into binary or scalar values describing node connectedness. In our paper instead, links are reverted to hypostatic relationships between entities with descriptional attributes. We propose GCN-LASE (GCN with Link Attributes and Sampling Estimation), a novel GCN model taking both node and link attributes as inputs. To adequately captures the interactions between link and node attributes, their tensor product is used as neighbor features, based on which we define several graph kernels and further develop according architectures for LASE. Besides, to accelerate the training process, the sum of features in entire neighborhoods are estimated through Monte Carlo method, with novel sampling strategies designed for LASE to minimize the estimation variance. Our experiments show that LASE outperforms strong baselines over various graph datasets, and further experiments corroborate the informativeness of link attributes and our model's ability of adequately leveraging them.
SepNE: Bringing Separability to Network Embedding
Nov 14, 2018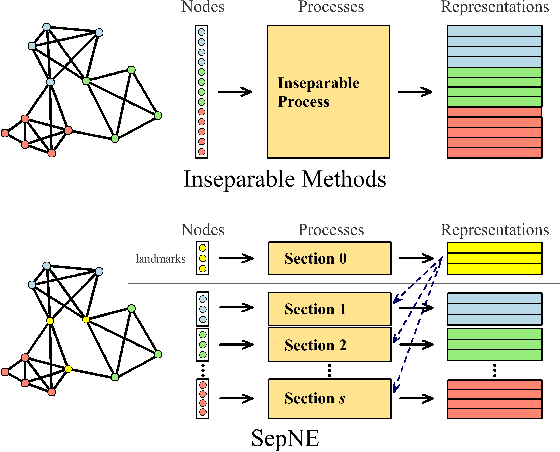
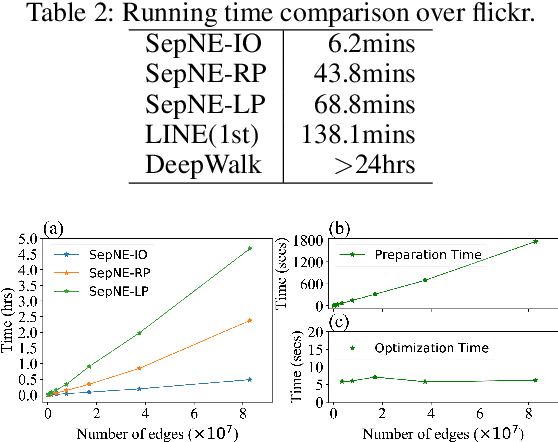
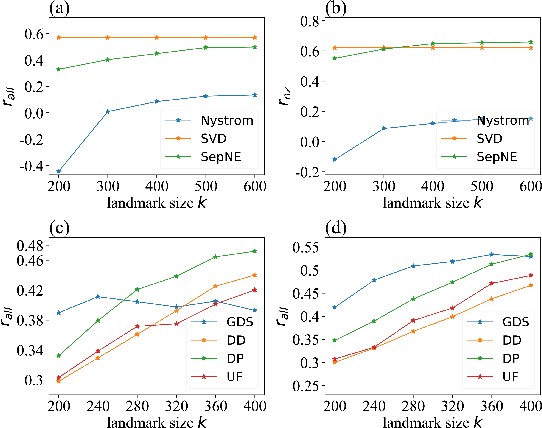
Many successful methods have been proposed for learning low dimensional representations on large-scale networks, while almost all existing methods are designed in inseparable processes, learning embeddings for entire networks even when only a small proportion of nodes are of interest. This leads to great inconvenience, especially on super-large or dynamic networks, where these methods become almost impossible to implement. In this paper, we formalize the problem of separated matrix factorization, based on which we elaborate a novel objective function that preserves both local and global information. We further propose SepNE, a simple and flexible network embedding algorithm which independently learns representations for different subsets of nodes in separated processes. By implementing separability, our algorithm reduces the redundant efforts to embed irrelevant nodes, yielding scalability to super-large networks, automatic implementation in distributed learning and further adaptations. We demonstrate the effectiveness of this approach on several real-world networks with different scales and subjects. With comparable accuracy, our approach significantly outperforms state-of-the-art baselines in running times on large networks.