 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSCALE: Self-uncertainty Conditioned Adaptive Looking and Execution for Vision-Language-Action Models
Feb 04, 2026Vision-Language-Action (VLA) models have emerged as a promising paradigm for general-purpose robotic control, with test-time scaling (TTS) gaining attention to enhance robustness beyond training. However, existing TTS methods for VLAs require additional training, verifiers, and multiple forward passes, making them impractical for deployment. Moreover, they intervene only at action decoding while keeping visual representations fixed-insufficient under perceptual ambiguity, where reconsidering how to perceive is as important as deciding what to do. To address these limitations, we propose SCALE, a simple inference strategy that jointly modulates visual perception and action based on 'self-uncertainty', inspired by uncertainty-driven exploration in Active Inference theory-requiring no additional training, no verifier, and only a single forward pass. SCALE broadens exploration in both perception and action under high uncertainty, while focusing on exploitation when confident-enabling adaptive execution across varying conditions. Experiments on simulated and real-world benchmarks demonstrate that SCALE improves state-of-the-art VLAs and outperforms existing TTS methods while maintaining single-pass efficiency.
BioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024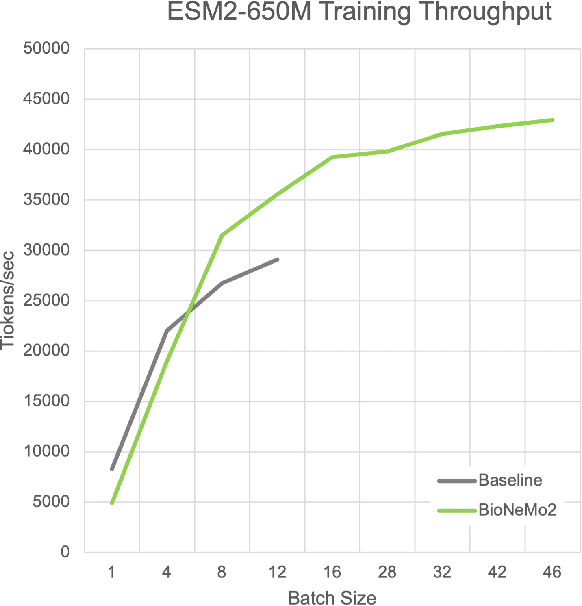
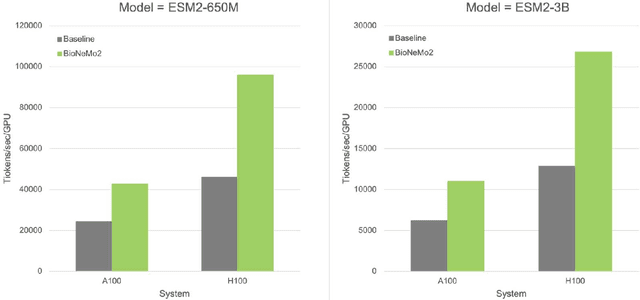
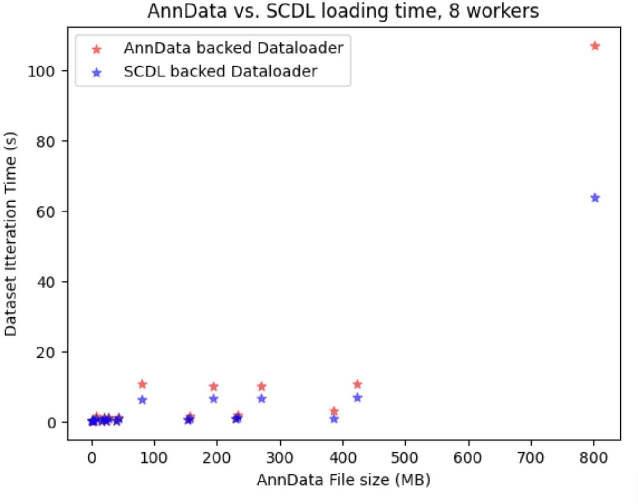
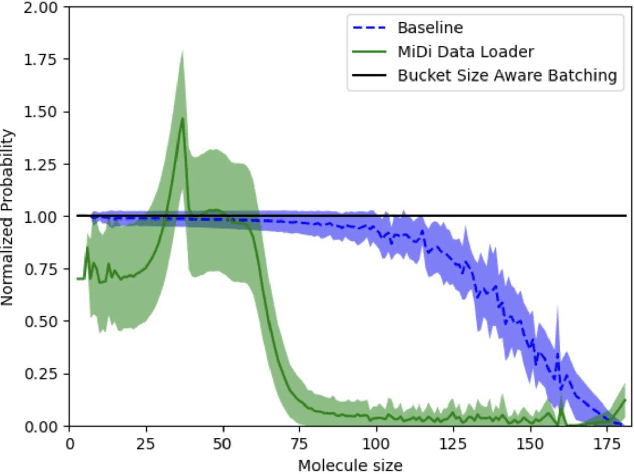
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
BOK-VQA: Bilingual Outside Knowledge-based Visual Question Answering via Graph Representation Pretraining
Jan 12, 2024The current research direction in generative models, such as the recently developed GPT4, aims to find relevant knowledge information for multimodal and multilingual inputs to provide answers. Under these research circumstances, the demand for multilingual evaluation of visual question answering (VQA) tasks, a representative task of multimodal systems, has increased. Accordingly, we propose a bilingual outside-knowledge VQA (BOK-VQA) dataset in this study that can be extended to multilingualism. The proposed data include 17K images, 17K question-answer pairs for both Korean and English and 280K instances of knowledge information related to question-answer content. We also present a framework that can effectively inject knowledge information into a VQA system by pretraining the knowledge information of BOK-VQA data in the form of graph embeddings. Finally, through in-depth analysis, we demonstrated the actual effect of the knowledge information contained in the constructed training data on VQA.
Solvent: A Framework for Protein Folding
Jul 31, 2023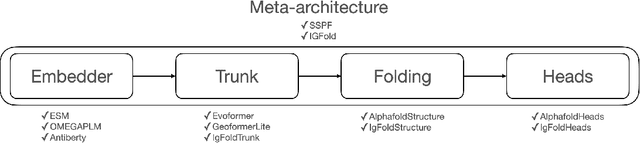

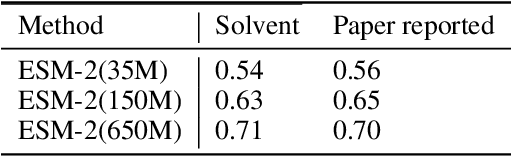
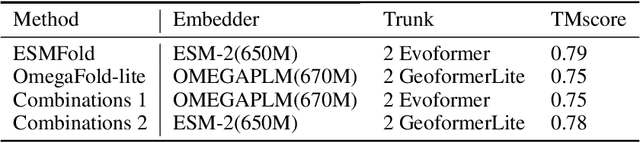
Consistency and reliability are crucial for conducting AI research. Many famous research fields, such as object detection, have been compared and validated with solid benchmark frameworks. After AlphaFold2, the protein folding task has entered a new phase, and many methods are proposed based on the component of AlphaFold2. The importance of a unified research framework in protein folding contains implementations and benchmarks to consistently and fairly compare various approaches. To achieve this, we present Solvent, a protein folding framework that supports significant components of state-of-the-art models in the manner of an off-the-shelf interface Solvent contains different models implemented in a unified codebase and supports training and evaluation for defined models on the same dataset. We benchmark well-known algorithms and their components and provide experiments that give helpful insights into the protein structure modeling field. We hope that Solvent will increase the reliability and consistency of proposed models and give efficiency in both speed and costs, resulting in acceleration on protein folding modeling research. The code is available at https://github.com/kakaobrain/solvent, and the project will continue to be developed.
ProtFIM: Fill-in-Middle Protein Sequence Design via Protein Language Models
Mar 29, 2023



Protein language models (pLMs), pre-trained via causal language modeling on protein sequences, have been a promising tool for protein sequence design. In real-world protein engineering, there are many cases where the amino acids in the middle of a protein sequence are optimized while maintaining other residues. Unfortunately, because of the left-to-right nature of pLMs, existing pLMs modify suffix residues by prompting prefix residues, which are insufficient for the infilling task that considers the whole surrounding context. To find the more effective pLMs for protein engineering, we design a new benchmark, Secondary structureE InFilling rEcoveRy, SEIFER, which approximates infilling sequence design scenarios. With the evaluation of existing models on the benchmark, we reveal the weakness of existing language models and show that language models trained via fill-in-middle transformation, called ProtFIM, are more appropriate for protein engineering. Also, we prove that ProtFIM generates protein sequences with decent protein representations through exhaustive experiments and visualizations.
Predictive models of RNA degradation through dual crowdsourcing
Oct 14, 2021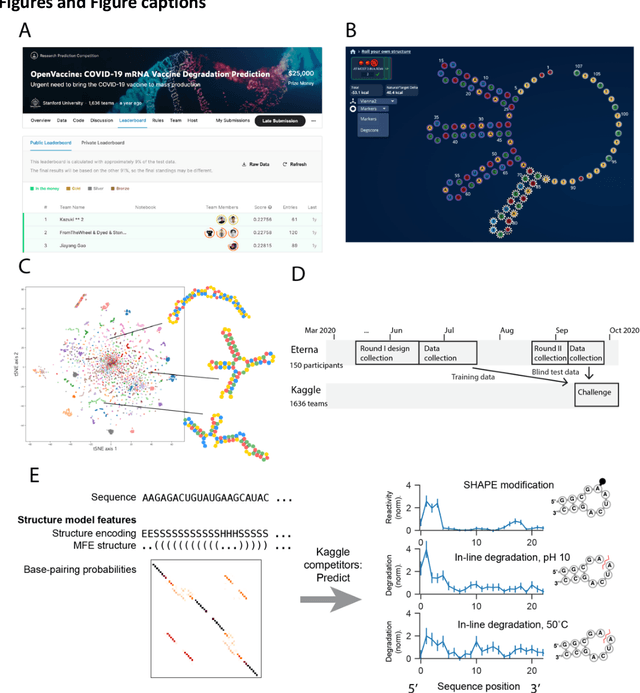
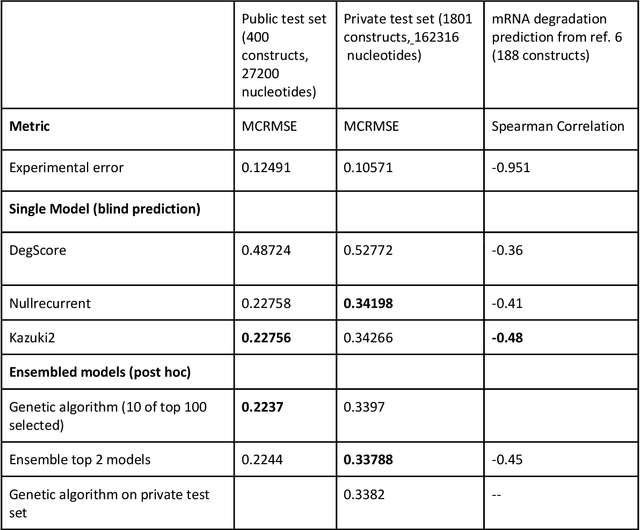
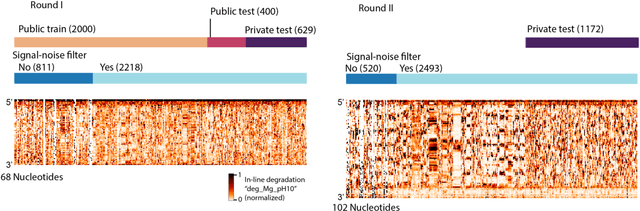
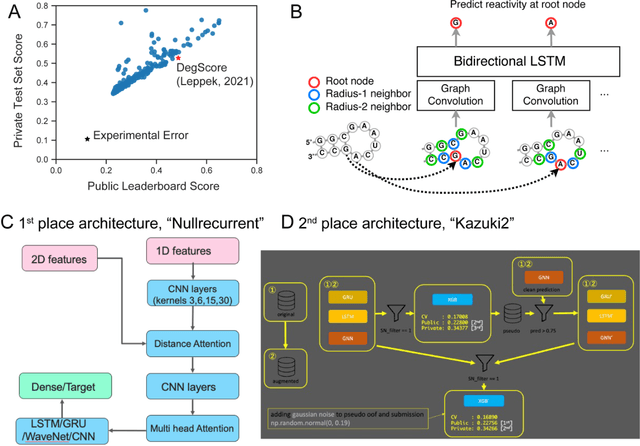
Messenger RNA-based medicines hold immense potential, as evidenced by their rapid deployment as COVID-19 vaccines. However, worldwide distribution of mRNA molecules has been limited by their thermostability, which is fundamentally limited by the intrinsic instability of RNA molecules to a chemical degradation reaction called in-line hydrolysis. Predicting the degradation of an RNA molecule is a key task in designing more stable RNA-based therapeutics. Here, we describe a crowdsourced machine learning competition ("Stanford OpenVaccine") on Kaggle, involving single-nucleotide resolution measurements on 6043 102-130-nucleotide diverse RNA constructs that were themselves solicited through crowdsourcing on the RNA design platform Eterna. The entire experiment was completed in less than 6 months. Winning models demonstrated test set errors that were better by 50% than the previous state-of-the-art DegScore model. Furthermore, these models generalized to blindly predicting orthogonal degradation data on much longer mRNA molecules (504-1588 nucleotides) with improved accuracy over DegScore and other models. Top teams integrated natural language processing architectures and data augmentation techniques with predictions from previous dynamic programming models for RNA secondary structure. These results indicate that such models are capable of representing in-line hydrolysis with excellent accuracy, supporting their use for designing stabilized messenger RNAs. The integration of two crowdsourcing platforms, one for data set creation and another for machine learning, may be fruitful for other urgent problems that demand scientific discovery on rapid timescales.
A community-powered search of machine learning strategy space to find NMR property prediction models
Aug 13, 2020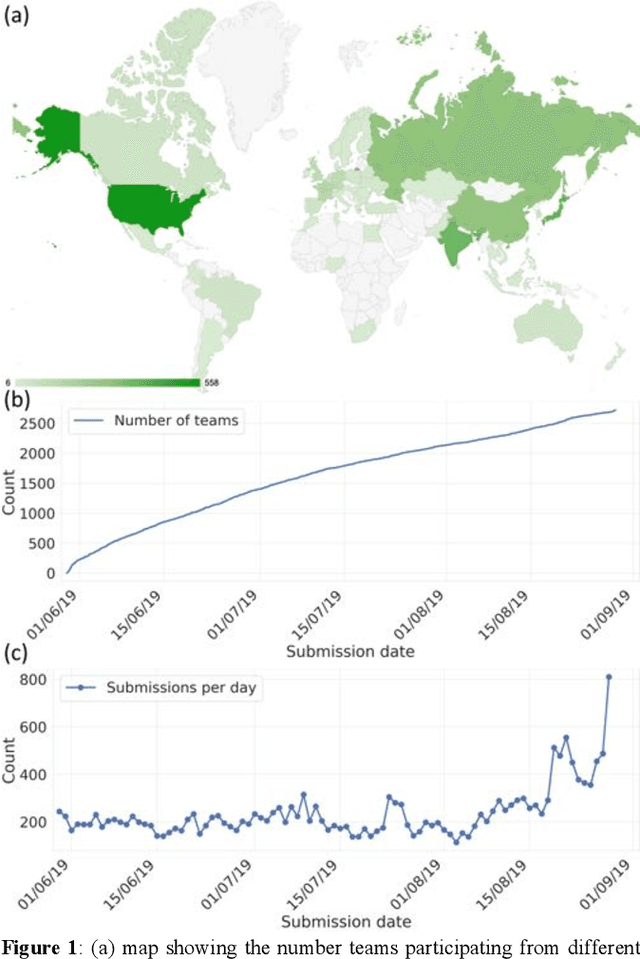
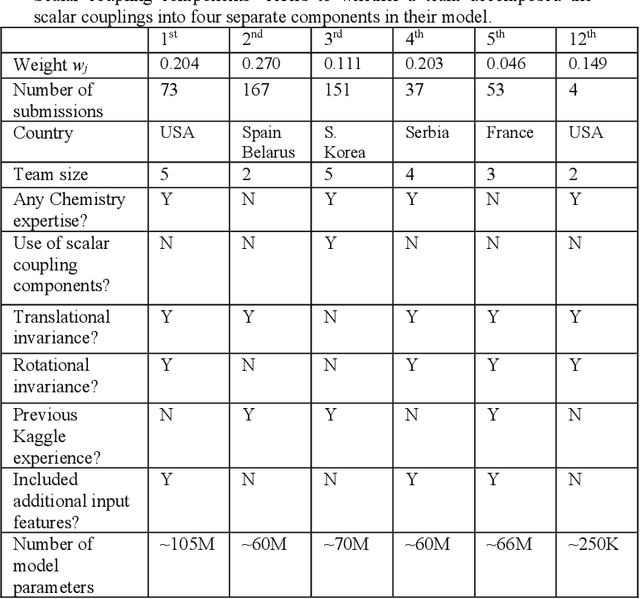
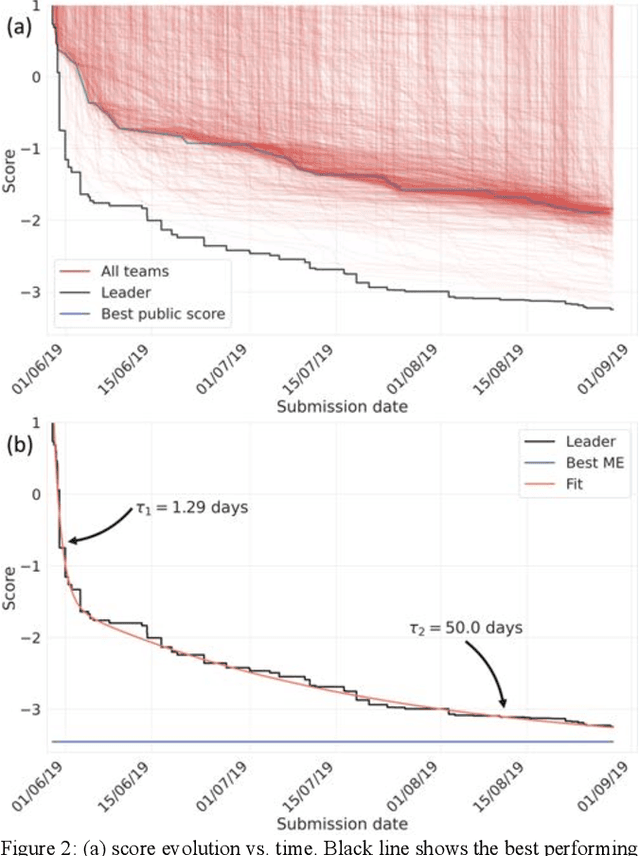
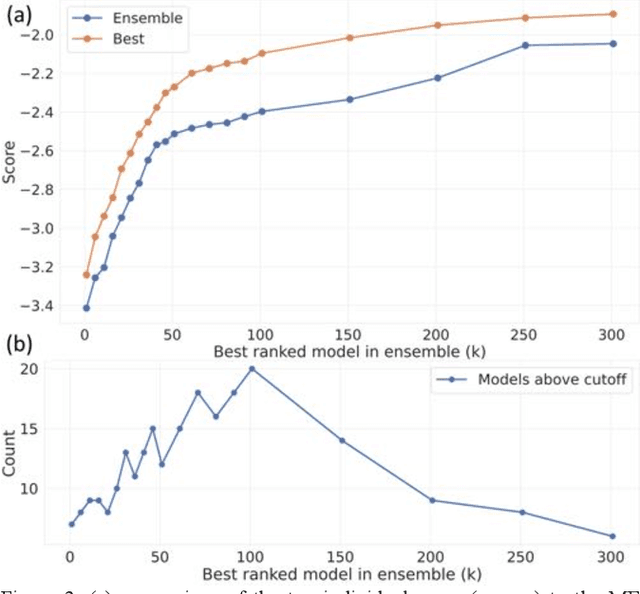
The rise of machine learning (ML) has created an explosion in the potential strategies for using data to make scientific predictions. For physical scientists wishing to apply ML strategies to a particular domain, it can be difficult to assess in advance what strategy to adopt within a vast space of possibilities. Here we outline the results of an online community-powered effort to swarm search the space of ML strategies and develop algorithms for predicting atomic-pairwise nuclear magnetic resonance (NMR) properties in molecules. Using an open-source dataset, we worked with Kaggle to design and host a 3-month competition which received 47,800 ML model predictions from 2,700 teams in 84 countries. Within 3 weeks, the Kaggle community produced models with comparable accuracy to our best previously published "in-house" efforts. A meta-ensemble model constructed as a linear combination of the top predictions has a prediction accuracy which exceeds that of any individual model, 7-19x better than our previous state-of-the-art. The results highlight the potential of transformer architectures for predicting quantum mechanical (QM) molecular properties.