 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSolvent: A Framework for Protein Folding
Jul 31, 2023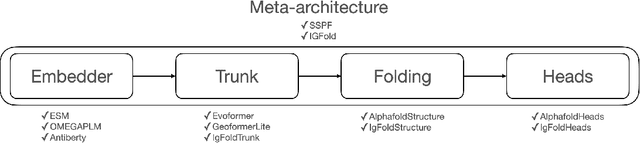

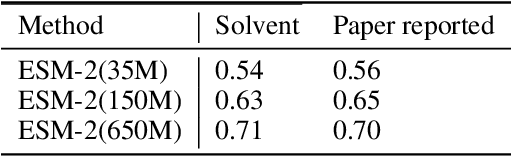
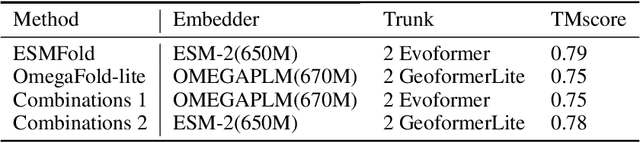
Consistency and reliability are crucial for conducting AI research. Many famous research fields, such as object detection, have been compared and validated with solid benchmark frameworks. After AlphaFold2, the protein folding task has entered a new phase, and many methods are proposed based on the component of AlphaFold2. The importance of a unified research framework in protein folding contains implementations and benchmarks to consistently and fairly compare various approaches. To achieve this, we present Solvent, a protein folding framework that supports significant components of state-of-the-art models in the manner of an off-the-shelf interface Solvent contains different models implemented in a unified codebase and supports training and evaluation for defined models on the same dataset. We benchmark well-known algorithms and their components and provide experiments that give helpful insights into the protein structure modeling field. We hope that Solvent will increase the reliability and consistency of proposed models and give efficiency in both speed and costs, resulting in acceleration on protein folding modeling research. The code is available at https://github.com/kakaobrain/solvent, and the project will continue to be developed.
ProtFIM: Fill-in-Middle Protein Sequence Design via Protein Language Models
Mar 29, 2023



Protein language models (pLMs), pre-trained via causal language modeling on protein sequences, have been a promising tool for protein sequence design. In real-world protein engineering, there are many cases where the amino acids in the middle of a protein sequence are optimized while maintaining other residues. Unfortunately, because of the left-to-right nature of pLMs, existing pLMs modify suffix residues by prompting prefix residues, which are insufficient for the infilling task that considers the whole surrounding context. To find the more effective pLMs for protein engineering, we design a new benchmark, Secondary structureE InFilling rEcoveRy, SEIFER, which approximates infilling sequence design scenarios. With the evaluation of existing models on the benchmark, we reveal the weakness of existing language models and show that language models trained via fill-in-middle transformation, called ProtFIM, are more appropriate for protein engineering. Also, we prove that ProtFIM generates protein sequences with decent protein representations through exhaustive experiments and visualizations.