 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMitosis Detection in the Wild: Multi-Tumor and Context-Aware Generalization in the MIDOG 2025 Challenge
Jun 05, 2026Automated mitosis detection is a well-established task in computational pathology. While previous benchmarks focused on scanner-induced domain shift, clinical "real-world" application requires models to be robust across the vast variance to be expected in the histological landscape. The MItosis DOmain Generalization (MIDOG) 2025 challenge was designed to evaluate algorithmic performance across unprecedented biological and contextual diversity. We curated a test dataset of 365 cases, encompassing 12 distinct human, canine and feline tumor types, digitized across multiple scanning platforms. Moving beyond hand-selected hotspots, the challenge required detection also in random tissue areas (representative of the whole slide detection situation) and challenging areas (areas rich in hard negatives). In the second track, we introduced the classification of atypical mitotic figures (AMFs). There were 18 teams submitting to the detection track, with F1 scores ranging up to 0.740. In the AMF detection track, we had 21 submissions with balanced accuracy values up to 0.908. Our analysis reveals that while most models perform reliably in traditional hotspots, significant performance degradation occurs in challenging ROIs, where false positive rates tripled. Furthermore, performance varied significantly across the 12 tumor types, highlighting "blind spots" in current state-of-the-art architectures when encountering rare or highly pleomorphic malignancies. Moreover, we evaluated the effectiveness of ensembling and found a mean increases of 1.5 and 1.3 percentage points in F1 score and balanced accuracy, respectively. In contrast, TTA showed no relevant improvement. MIDOG 2025 demonstrates that "in the wild" mitosis detection remains a significant hurdle. The transition from hotspot-only evaluation to a multi-contextual framework provides a more realistic proxy for clinical reliability.
LEMON: a foundation model for nuclear morphology in Computational Pathology
Mar 26, 2026Computational pathology relies on effective representation learning to support cancer research and precision medicine. Although self-supervised learning has driven major progress at the patch and whole-slide image levels, representation learning at the single-cell level remains comparatively underexplored, despite its importance for characterizing cell types and cellular phenotypes. We introduce LEMON (Learning Embeddings from Morphology Of Nuclei), a self-supervised foundation model for scalable single-cell image representation learning. Trained on millions of cell images from diverse tissues and cancer types, LEMON learns robust and versatile morphological representations that support large-scale single-cell analyses in pathology. We evaluate LEMON on five benchmark datasets across a range of prediction tasks and show that it provides strong performance, highlighting its potential as a new paradigm for cell-level computational pathology. Model weights are available at https://huggingface.co/aliceblondel/LEMON.
Efficient Fine-Tuning of DINOv3 Pretrained on Natural Images for Atypical Mitotic Figure Classification in MIDOG 2025
Aug 28, 2025Atypical mitotic figures (AMFs) are markers of abnormal cell division associated with poor prognosis, yet their detection remains difficult due to low prevalence, subtle morphology, and inter-observer variability. The MIDOG 2025 challenge introduces a benchmark for AMF classification across multiple domains. In this work, we evaluate the recently published DINOv3-H+ vision transformer, pretrained on natural images, which we fine-tuned using low-rank adaptation (LoRA, 650k trainable parameters) and extensive augmentation. Despite the domain gap, DINOv3 transfers effectively to histopathology, achieving a balanced accuracy of 0.8871 on the preliminary test set. These results highlight the robustness of DINOv3 pretraining and show that, when combined with parameter-efficient fine-tuning, it provides a strong baseline for atypical mitosis classification in MIDOG 2025.
MIPHEI-ViT: Multiplex Immunofluorescence Prediction from H&E Images using ViT Foundation Models
May 15, 2025Histopathological analysis is a cornerstone of cancer diagnosis, with Hematoxylin and Eosin (H&E) staining routinely acquired for every patient to visualize cell morphology and tissue architecture. On the other hand, multiplex immunofluorescence (mIF) enables more precise cell type identification via proteomic markers, but has yet to achieve widespread clinical adoption due to cost and logistical constraints. To bridge this gap, we introduce MIPHEI (Multiplex Immunofluorescence Prediction from H&E), a U-Net-inspired architecture that integrates state-of-the-art ViT foundation models as encoders to predict mIF signals from H&E images. MIPHEI targets a comprehensive panel of markers spanning nuclear content, immune lineages (T cells, B cells, myeloid), epithelium, stroma, vasculature, and proliferation. We train our model using the publicly available ORION dataset of restained H&E and mIF images from colorectal cancer tissue, and validate it on two independent datasets. MIPHEI achieves accurate cell-type classification from H&E alone, with F1 scores of 0.88 for Pan-CK, 0.57 for CD3e, 0.56 for SMA, 0.36 for CD68, and 0.30 for CD20, substantially outperforming both a state-of-the-art baseline and a random classifier for most markers. Our results indicate that our model effectively captures the complex relationships between nuclear morphologies in their tissue context, as visible in H&E images and molecular markers defining specific cell types. MIPHEI offers a promising step toward enabling cell-type-aware analysis of large-scale H&E datasets, in view of uncovering relationships between spatial cellular organization and patient outcomes.
UWB Anchor Based Localization of a Planetary Rover
Apr 10, 2025



Localization of an autonomous mobile robot during planetary exploration is challenging due to the unknown terrain, the difficult lighting conditions and the lack of any global reference such as satellite navigation systems. We present a novel approach for robot localization based on ultra-wideband (UWB) technology. The robot sets up its own reference coordinate system by distributing UWB anchor nodes in the environment via a rocket-propelled launcher system. This allows the creation of a localization space in which UWB measurements are employed to supplement traditional SLAM-based techniques. The system was developed for our involvement in the ESA-ESRIC challenge 2021 and the AMADEE-24, an analog Mars simulation in Armenia by the Austrian Space Forum (\"OWF).
Interpretable Embeddings for Segmentation-Free Single-Cell Analysis in Multiplex Imaging
Nov 02, 2024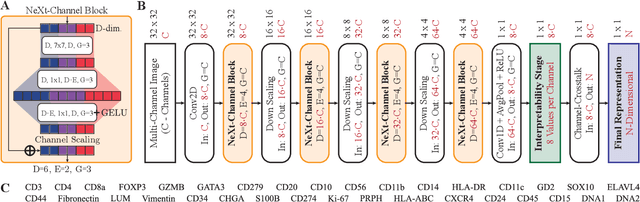

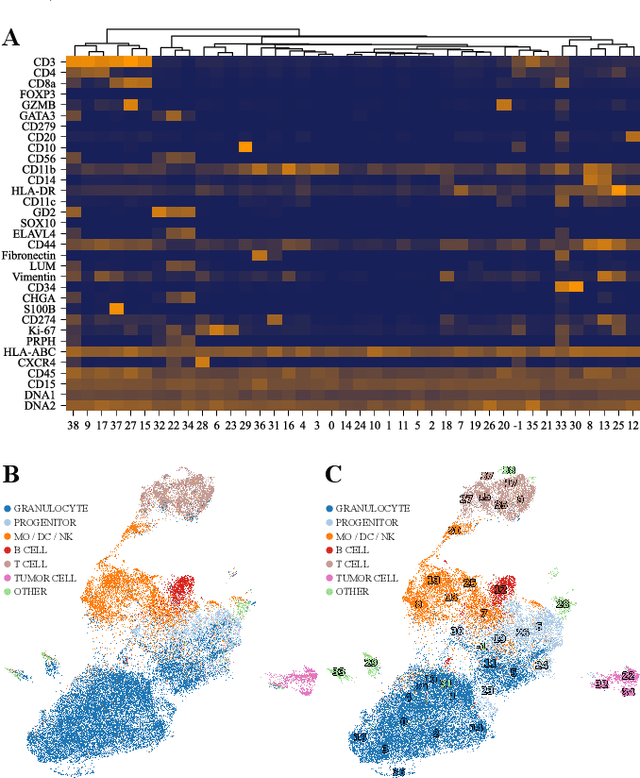
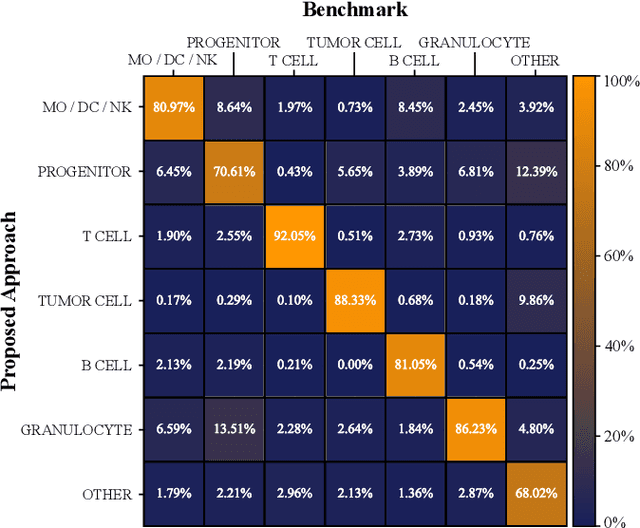
Multiplex Imaging (MI) enables the simultaneous visualization of multiple biological markers in separate imaging channels at subcellular resolution, providing valuable insights into cell-type heterogeneity and spatial organization. However, current computational pipelines rely on cell segmentation algorithms, which require laborious fine-tuning and can introduce downstream errors due to inaccurate single-cell representations. We propose a segmentation-free deep learning approach that leverages grouped convolutions to learn interpretable embedded features from each imaging channel, enabling robust cell-type identification without manual feature selection. Validated on an Imaging Mass Cytometry dataset of 1.8 million cells from neuroblastoma patients, our method enables the accurate identification of known cell types, showcasing its scalability and suitability for high-dimensional MI data.
Simple and Efficient Confidence Score for Grading Whole Slide Images
Mar 08, 2023



Grading precancerous lesions on whole slide images is a challenging task: the continuous space of morphological phenotypes makes clear-cut decisions between different grades often difficult, leading to low inter- and intra-rater agreements. More and more Artificial Intelligence (AI) algorithms are developed to help pathologists perform and standardize their diagnosis. However, those models can render their prediction without consideration of the ambiguity of the classes and can fail without notice which prevent their wider acceptance in a clinical context. In this paper, we propose a new score to measure the confidence of AI models in grading tasks. Our confidence score is specifically adapted to ordinal output variables, is versatile and does not require extra training or additional inferences nor particular architecture changes. Comparison to other popular techniques such as Monte Carlo Dropout and deep ensembles shows that our method provides state-of-the art results, while being simpler, more versatile and less computationally intensive. The score is also easily interpretable and consistent with real life hesitations of pathologists. We show that the score is capable of accurately identifying mispredicted slides and that accuracy for high confidence decisions is significantly higher than for low-confidence decisions (gap in AUC of 17.1% on the test set). We believe that the proposed confidence score could be leveraged by pathologists directly in their workflow and assist them on difficult tasks such as grading precancerous lesions.
PointFISH -- learning point cloud representations for RNA localization patterns
Feb 21, 2023Subcellular RNA localization is a critical mechanism for the spatial control of gene expression. Its mechanism and precise functional role is not yet very well understood. Single Molecule Fluorescence in Situ Hybridization (smFISH) images allow for the detection of individual RNA molecules with subcellular accuracy. In return, smFISH requires robust methods to quantify and classify RNA spatial distribution. Here, we present PointFISH, a novel computational approach for the recognition of RNA localization patterns. PointFISH is an attention-based network for computing continuous vector representations of RNA point clouds. Trained on simulations only, it can directly process extracted coordinates from experimental smFISH images. The resulting embedding allows scalable and flexible spatial transcriptomics analysis and matches performance of hand-crafted pipelines.
Learning with minimal effort: leveraging in silico labeling for cell and nucleus segmentation
Jan 10, 2023
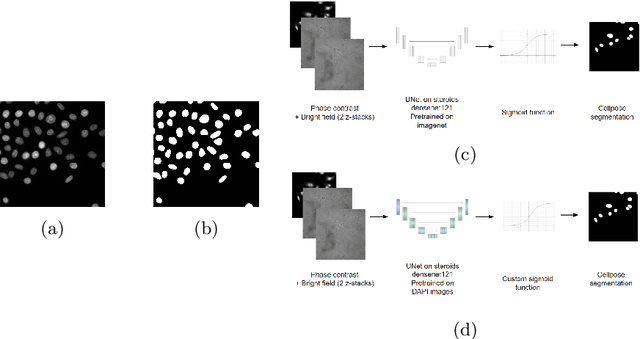


Deep learning provides us with powerful methods to perform nucleus or cell segmentation with unprecedented quality. However, these methods usually require large training sets of manually annotated images, which are tedious and expensive to generate. In this paper we propose to use In Silico Labeling (ISL) as a pretraining scheme for segmentation tasks. The strategy is to acquire label-free microscopy images (such as bright-field or phase contrast) along fluorescently labeled images (such as DAPI or CellMask). We then train a model to predict the fluorescently labeled images from the label-free microscopy images. By comparing segmentation performance across several training set sizes, we show that such a scheme can dramatically reduce the number of required annotations.
Giga-SSL: Self-Supervised Learning for Gigapixel Images
Dec 06, 2022



Whole slide images (WSI) are microscopy images of stained tissue slides routinely prepared for diagnosis and treatment selection in medical practice. WSI are very large (gigapixel size) and complex (made of up to millions of cells). The current state-of-the-art (SoTA) approach to classify WSI subdivides them into tiles, encodes them by pre-trained networks and applies Multiple Instance Learning (MIL) to train for specific downstream tasks. However, annotated datasets are often small, typically a few hundred to a few thousand WSI, which may cause overfitting and underperforming models. Conversely, the number of unannotated WSI is ever increasing, with datasets of tens of thousands (soon to be millions) of images available. While it has been previously proposed to use these unannotated data to identify suitable tile representations by self-supervised learning (SSL), downstream classification tasks still require full supervision because parts of the MIL architecture is not trained during tile level SSL pre-training. Here, we propose a strategy of slide level SSL to leverage the large number of WSI without annotations to infer powerful slide representations. Applying our method to The Cancer-Genome Atlas, one of the most widely used data resources in cancer research (16 TB image data), we are able to downsize the dataset to 23 MB without any loss in predictive power: we show that a linear classifier trained on top of these embeddings maintains or improves previous SoTA performances on various benchmark WSI classification tasks. Finally, we observe that training a classifier on these representations with tiny datasets (e.g. 50 slides) improved performances over SoTA by an average of +6.3 AUC points over all downstream tasks.