 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePreserving Marker Specificity with Lightweight Channel-Independent Representation Learning
Dec 17, 2025Multiplexed tissue imaging measures dozens of protein markers per cell, yet most deep learning models still apply early channel fusion, assuming shared structure across markers. We investigate whether preserving marker independence, combined with deliberately shallow architectures, provides a more suitable inductive bias for self-supervised representation learning in multiplex data than increasing model scale. Using a Hodgkin lymphoma CODEX dataset with 145,000 cells and 49 markers, we compare standard early-fusion CNNs with channel-separated architectures, including a marker-aware baseline and our novel shallow Channel-Independent Model (CIM-S) with 5.5K parameters. After contrastive pretraining and linear evaluation, early-fusion models show limited ability to retain marker-specific information and struggle particularly with rare-cell discrimination. Channel-independent architectures, and CIM-S in particular, achieve substantially stronger representations despite their compact size. These findings are consistent across multiple self-supervised frameworks, remain stable across augmentation settings, and are reproducible across both the 49-marker and reduced 18-marker settings. These results show that lightweight, channel-independent architectures can match or surpass deep early-fusion CNNs and foundation models for multiplex representation learning. Code is available at https://github.com/SimonBon/CIM-S.
Interpretable Embeddings for Segmentation-Free Single-Cell Analysis in Multiplex Imaging
Nov 02, 2024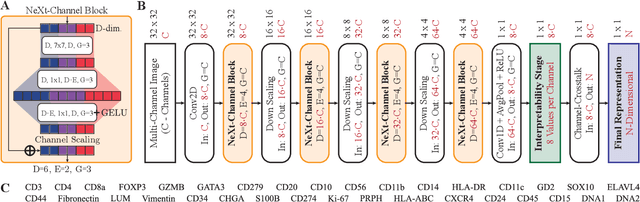

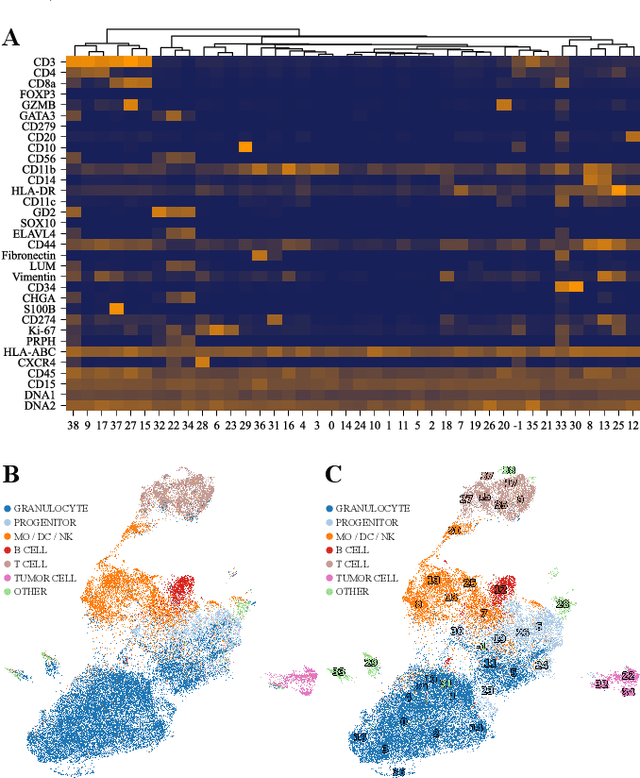
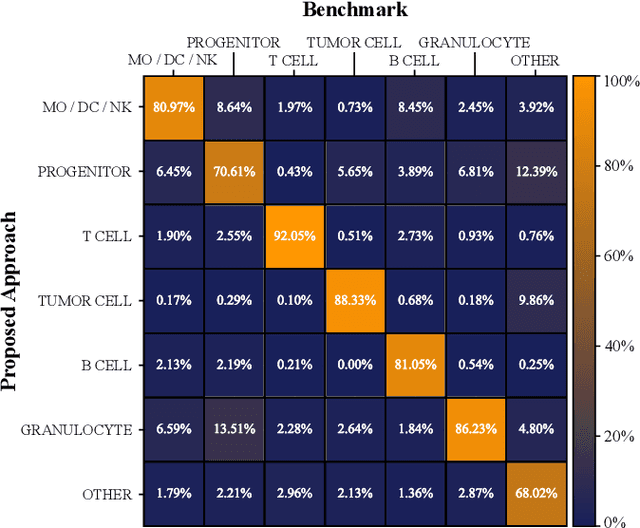
Multiplex Imaging (MI) enables the simultaneous visualization of multiple biological markers in separate imaging channels at subcellular resolution, providing valuable insights into cell-type heterogeneity and spatial organization. However, current computational pipelines rely on cell segmentation algorithms, which require laborious fine-tuning and can introduce downstream errors due to inaccurate single-cell representations. We propose a segmentation-free deep learning approach that leverages grouped convolutions to learn interpretable embedded features from each imaging channel, enabling robust cell-type identification without manual feature selection. Validated on an Imaging Mass Cytometry dataset of 1.8 million cells from neuroblastoma patients, our method enables the accurate identification of known cell types, showcasing its scalability and suitability for high-dimensional MI data.
FISHing in Uncertainty: Synthetic Contrastive Learning for Genetic Aberration Detection
Nov 01, 2024Detecting genetic aberrations is crucial in cancer diagnosis, typically through fluorescence in situ hybridization (FISH). However, existing FISH image classification methods face challenges due to signal variability, the need for costly manual annotations and fail to adequately address the intrinsic uncertainty. We introduce a novel approach that leverages synthetic images to eliminate the requirement for manual annotations and utilizes a joint contrastive and classification objective for training to account for inter-class variation effectively. We demonstrate the superior generalization capabilities and uncertainty calibration of our method, which is trained on synthetic data, by testing it on a manually annotated dataset of real-world FISH images. Our model offers superior calibration in terms of classification accuracy and uncertainty quantification with a classification accuracy of 96.7% among the 50% most certain cases. The presented end-to-end method reduces the demands on personnel and time and improves the diagnostic workflow due to its accuracy and adaptability. All code and data is publicly accessible at: https://github.com/SimonBon/FISHing
Deep Learning architectures for generalized immunofluorescence based nuclear image segmentation
Jul 30, 2019
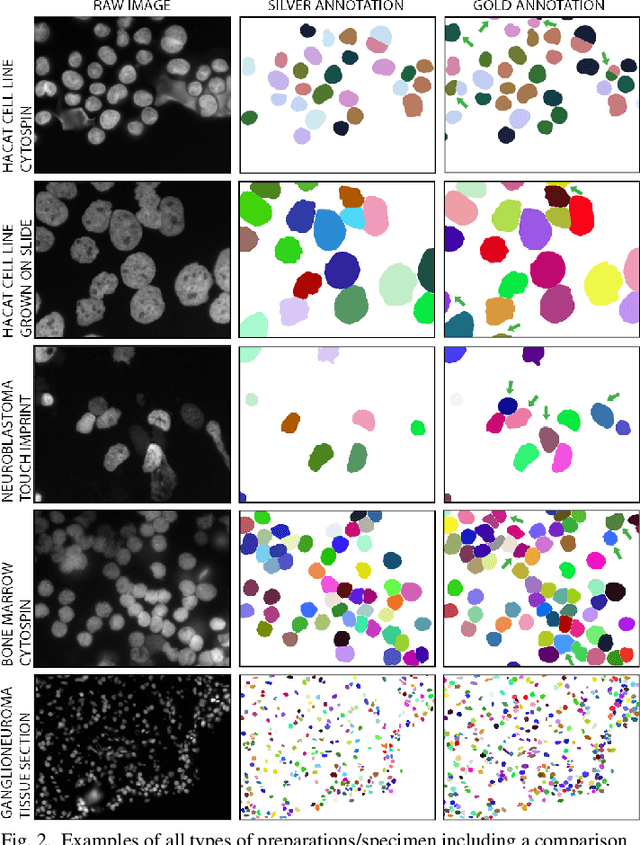
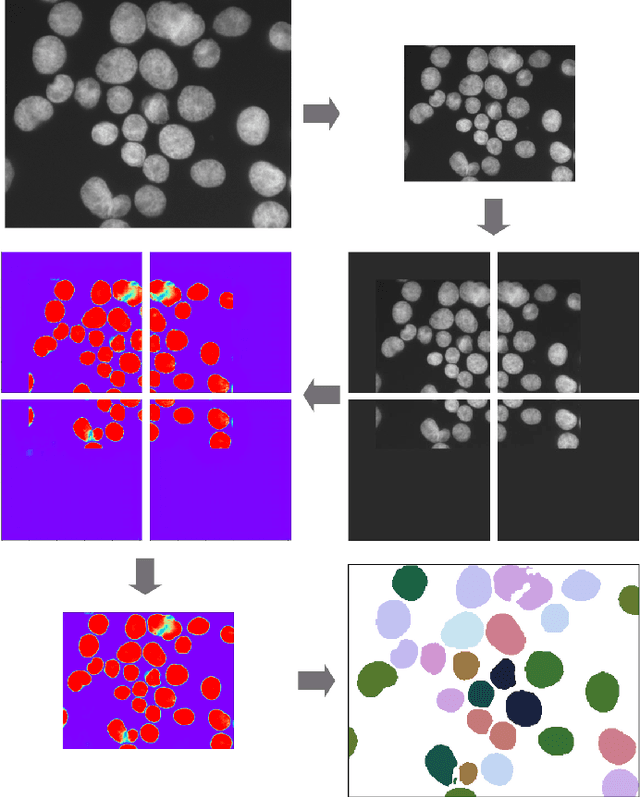

Separating and labeling each instance of a nucleus (instance-aware segmentation) is the key challenge in segmenting single cell nuclei on fluorescence microscopy images. Deep Neural Networks can learn the implicit transformation of a nuclear image into a probability map indicating the class membership of each pixel (nucleus or background), but the use of post-processing steps to turn the probability map into a labeled object mask is error-prone. This especially accounts for nuclear images of tissue sections and nuclear images across varying tissue preparations. In this work, we aim to evaluate the performance of state-of-the-art deep learning architectures to segment nuclei in fluorescence images of various tissue origins and sample preparation types without post-processing. We compare architectures that operate on pixel to pixel translation and an architecture that operates on object detection and subsequent locally applied segmentation. In addition, we propose a novel strategy to create artificial images to extend the training set. We evaluate the influence of ground truth annotation quality, image scale and segmentation complexity on segmentation performance. Results show that three out of four deep learning architectures (U-Net, U-Net with ResNet34 backbone, Mask R-CNN) can segment fluorescent nuclear images on most of the sample preparation types and tissue origins with satisfactory segmentation performance. Mask R-CNN, an architecture designed to address instance aware segmentation tasks, outperforms other architectures. Equal nuclear mean size, consistent nuclear annotations and the use of artificially generated images result in overall acceptable precision and recall across different tissues and sample preparation types.
Deep SNP: An End-to-end Deep Neural Network with Attention-based Localization for Break-point Detection in SNP Array Genomic data
Jun 22, 2018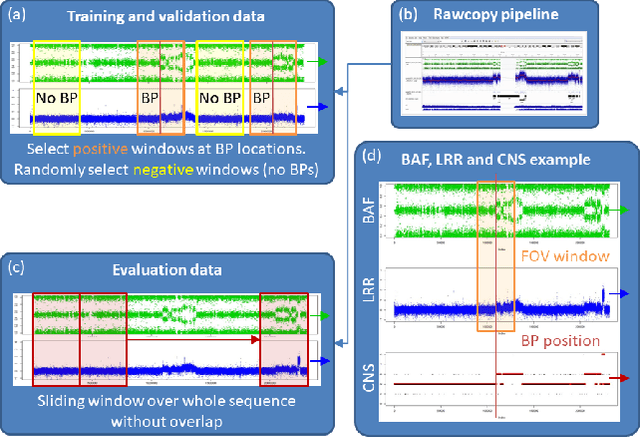

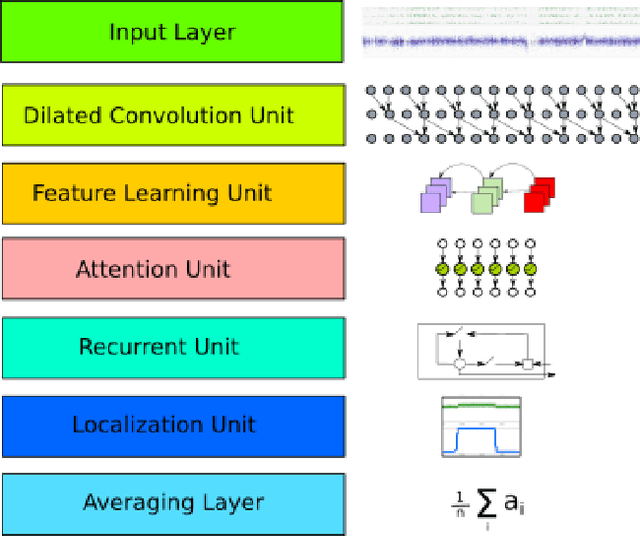
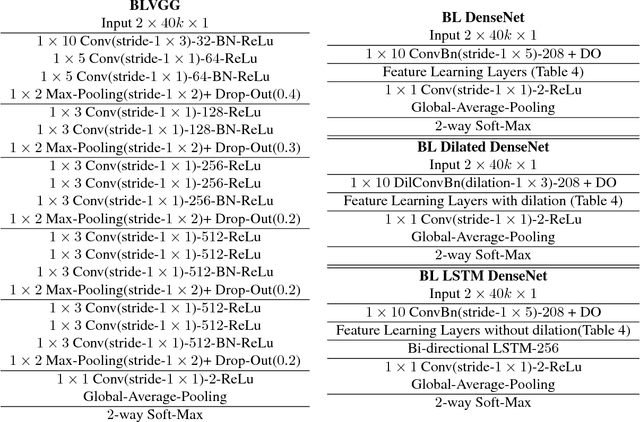
Diagnosis and risk stratification of cancer and many other diseases require the detection of genomic breakpoints as a prerequisite of calling copy number alterations (CNA). This, however, is still challenging and requires time-consuming manual curation. As deep-learning methods outperformed classical state-of-the-art algorithms in various domains and have also been successfully applied to life science problems including medicine and biology, we here propose Deep SNP, a novel Deep Neural Network to learn from genomic data. Specifically, we used a manually curated dataset from 12 genomic single nucleotide polymorphism array (SNPa) profiles as truth-set and aimed at predicting the presence or absence of genomic breakpoints, an indicator of structural chromosomal variations, in windows of 40,000 probes. We compare our results with well-known neural network models as well as Rawcopy though this tool is designed to predict breakpoints and in addition genomic segments with high sensitivity. We show, that Deep SNP is capable of successfully predicting the presence or absence of a breakpoint in large genomic windows and outperforms state-of-the-art neural network models. Qualitative examples suggest that integration of a localization unit may enable breakpoint detection and prediction of genomic segments, even if the breakpoint coordinates were not provided for network training. These results warrant further evaluation of DeepSNP for breakpoint localization and subsequent calling of genomic segments.