 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAgents' Last Exam
Jun 03, 2026Recent AI systems have achieved strong results on a wide range of benchmarks, yet these gains have not translated into economically meaningful deployment across many professional domains. We argue that this gap is largely an evaluation problem: widely used benchmarks lack sustained performance measurement on real and economically valuable workflows. This paper introduces Agents' Last Exam (ALE), a benchmark designed to evaluate AI agents on long-horizon, economically valuable, real-world tasks with verifiable outcomes. Developed in collaboration with 250+ industry experts, ALE covers non-physical industries defined with reference to O*NET / SOC 2018 (the U.S. federal occupational taxonomy). It is organized around a task taxonomy with 55 subfields grouped into 13 industry clusters covering 1K+ tasks. Current results show that the hardest tier remains far from saturated: across mainstream harness and backbone configurations, the average full pass rate is 2.6%. ALE is designed as a living benchmark: its task pool grows continuously as new workflows and industries are onboarded. More broadly, ALE is intended not merely as another leaderboard, but as an instrument for closing the gap between benchmark success and GDP-relevant impact.
Integrating Pathology Foundation Models and Spatial Transcriptomics for Cellular Decomposition from Histology Images
Jul 09, 2025The rapid development of digital pathology and modern deep learning has facilitated the emergence of pathology foundation models that are expected to solve general pathology problems under various disease conditions in one unified model, with or without fine-tuning. In parallel, spatial transcriptomics has emerged as a transformative technology that enables the profiling of gene expression on hematoxylin and eosin (H&E) stained histology images. Spatial transcriptomics unlocks the unprecedented opportunity to dive into existing histology images at a more granular, cellular level. In this work, we propose a lightweight and training-efficient approach to predict cellular composition directly from H&E-stained histology images by leveraging information-enriched feature embeddings extracted from pre-trained pathology foundation models. By training a lightweight multi-layer perceptron (MLP) regressor on cell-type abundances derived via cell2location, our method efficiently distills knowledge from pathology foundation models and demonstrates the ability to accurately predict cell-type compositions from histology images, without physically performing the costly spatial transcriptomics. Our method demonstrates competitive performance compared to existing methods such as Hist2Cell, while significantly reducing computational complexity.
Mimicking or Reasoning: Rethinking Multi-Modal In-Context Learning in Vision-Language Models
Jun 09, 2025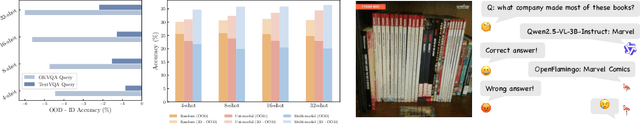

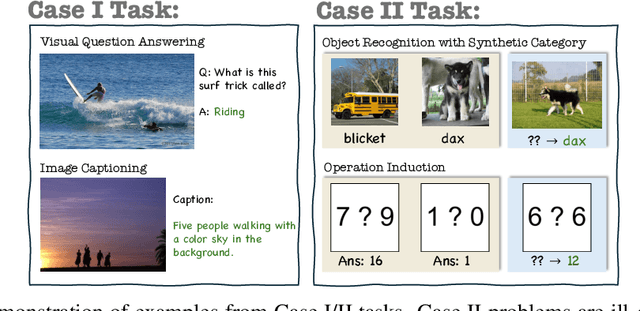
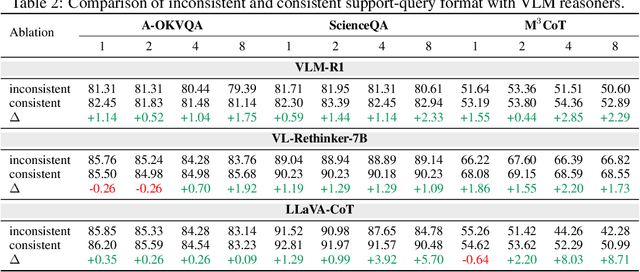
Vision-language models (VLMs) are widely assumed to exhibit in-context learning (ICL), a property similar to that of their language-only counterparts. While recent work suggests VLMs can perform multimodal ICL (MM-ICL), studies show they often rely on shallow heuristics -- such as copying or majority voting -- rather than true task understanding. We revisit this assumption by evaluating VLMs under distribution shifts, where support examples come from a dataset different from the query. Surprisingly, performance often degrades with more demonstrations, and models tend to copy answers rather than learn from them. To investigate further, we propose a new MM-ICL with Reasoning pipeline that augments each demonstration with a generated rationale alongside the answer. We conduct extensive and comprehensive experiments on both perception- and reasoning-required datasets with open-source VLMs ranging from 3B to 72B and proprietary models such as Gemini 2.0. We conduct controlled studies varying shot count, retrieval method, rationale quality, and distribution. Our results show limited performance sensitivity across these factors, suggesting that current VLMs do not effectively utilize demonstration-level information as intended in MM-ICL.
Diffuse Everything: Multimodal Diffusion Models on Arbitrary State Spaces
Jun 09, 2025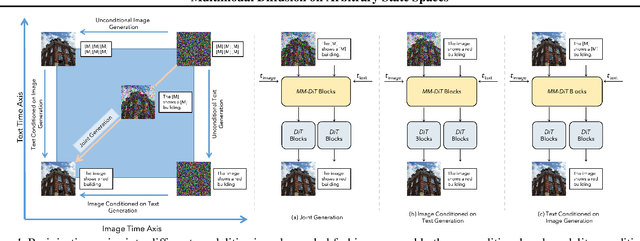
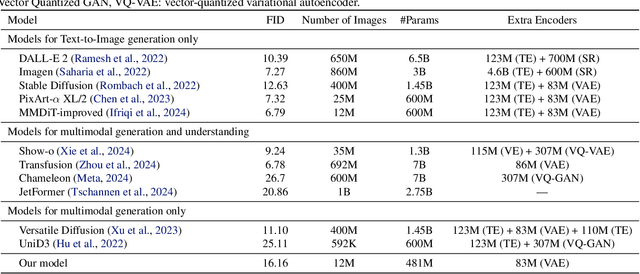
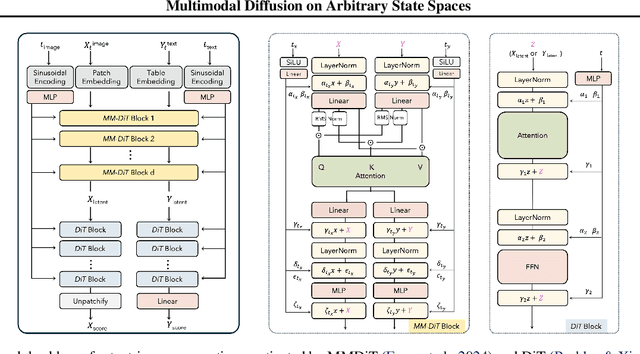
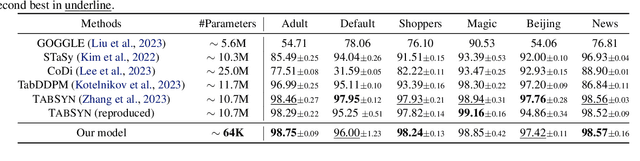
Diffusion models have demonstrated remarkable performance in generating unimodal data across various tasks, including image, video, and text generation. On the contrary, the joint generation of multimodal data through diffusion models is still in the early stages of exploration. Existing approaches heavily rely on external preprocessing protocols, such as tokenizers and variational autoencoders, to harmonize varied data representations into a unified, unimodal format. This process heavily demands the high accuracy of encoders and decoders, which can be problematic for applications with limited data. To lift this restriction, we propose a novel framework for building multimodal diffusion models on arbitrary state spaces, enabling native generation of coupled data across different modalities. By introducing an innovative decoupled noise schedule for each modality, we enable both unconditional and modality-conditioned generation within a single model simultaneously. We empirically validate our approach for text-image generation and mixed-type tabular data synthesis, demonstrating that it achieves competitive performance.
Diffusion Generative Modeling for Spatially Resolved Gene Expression Inference from Histology Images
Jan 26, 2025



Spatial Transcriptomics (ST) allows a high-resolution measurement of RNA sequence abundance by systematically connecting cell morphology depicted in Hematoxylin and Eosin (H&E) stained histology images to spatially resolved gene expressions. ST is a time-consuming, expensive yet powerful experimental technique that provides new opportunities to understand cancer mechanisms at a fine-grained molecular level, which is critical for uncovering new approaches for disease diagnosis and treatments. Here, we present $\textbf{Stem}$ ($\textbf{S}$pa$\textbf{T}$ially resolved gene $\textbf{E}$xpression inference with diffusion $\textbf{M}$odel), a novel computational tool that leverages a conditional diffusion generative model to enable in silico gene expression inference from H&E stained images. Through better capturing the inherent stochasticity and heterogeneity in ST data, $\textbf{Stem}$ achieves state-of-the-art performance on spatial gene expression prediction and generates biologically meaningful gene profiles for new H&E stained images at test time. We evaluate the proposed algorithm on datasets with various tissue sources and sequencing platforms, where it demonstrates clear improvement over existing approaches. $\textbf{Stem}$ generates high-fidelity gene expression predictions that share similar gene variation levels as ground truth data, suggesting that our method preserves the underlying biological heterogeneity. Our proposed pipeline opens up the possibility of analyzing existing, easily accessible H&E stained histology images from a genomics point of view without physically performing gene expression profiling and empowers potential biological discovery from H&E stained histology images.
Cell Spatial Analysis in Crohn's Disease: Unveiling Local Cell Arrangement Pattern with Graph-based Signatures
Aug 20, 2023Crohn's disease (CD) is a chronic and relapsing inflammatory condition that affects segments of the gastrointestinal tract. CD activity is determined by histological findings, particularly the density of neutrophils observed on Hematoxylin and Eosin stains (H&E) imaging. However, understanding the broader morphometry and local cell arrangement beyond cell counting and tissue morphology remains challenging. To address this, we characterize six distinct cell types from H&E images and develop a novel approach for the local spatial signature of each cell. Specifically, we create a 10-cell neighborhood matrix, representing neighboring cell arrangements for each individual cell. Utilizing t-SNE for non-linear spatial projection in scatter-plot and Kernel Density Estimation contour-plot formats, our study examines patterns of differences in the cellular environment associated with the odds ratio of spatial patterns between active CD and control groups. This analysis is based on data collected at the two research institutes. The findings reveal heterogeneous nearest-neighbor patterns, signifying distinct tendencies of cell clustering, with a particular focus on the rectum region. These variations underscore the impact of data heterogeneity on cell spatial arrangements in CD patients. Moreover, the spatial distribution disparities between the two research sites highlight the significance of collaborative efforts among healthcare organizations. All research analysis pipeline tools are available at https://github.com/MASILab/cellNN.