 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Neural Network based Hierarchy-Aware Embeddings of Knowledge Graphs: Applications to Yeast Phenotype Prediction
May 05, 2026We present a method for finding hierarchy-aware embeddings of knowledge graphs (KGs) using graph neural networks (GNNs) enriched with a semantic loss derived from underlying ontologies. This method yields embeddings that better reflect domain knowledge. To demonstrate their utility, we predict and interpret the effects of gene deletions in the yeast Saccharomyces cerevisiae and learn box embeddings for KGs in the absence of a prediction task. We further show how box embeddings can serve as the basis for evaluating KG revisions. Our yeast KG is constructed from community databases and ontology terms. Low-dimensional box embeddings combined with GNNs are used to predict cell growth for double gene knockouts. Over 10-fold cross validation, these predictions have a mean $R^2$~score~of~0.360, significantly higher than baseline comparisons, demonstrating that high-level qualitative knowledge is informative about experimental outcomes. Incorporating semantic loss terms in the training of the models improves their predictive performance ($R^2$=0.377) by aligning embeddings with ontology structure. This shows that class hierarchies from ontologies can be exploited for quantitative prediction. We also test the trained models on triple gene knockouts, showing they generalise to data beyond those seen in training. Additionally, by identifying co-occurring relations in the yeast KG important for the cell-growth predictions, we construct hypotheses about interacting traits in yeast. A biological experiment validates one such finding, revealing an association between inositol utilisation and osmotic stress resistance, highlighting the model's potential to guide biological discovery.
Compressing Chemistry Reveals Functional Groups
Nov 07, 2025We introduce the first formal large-scale assessment of the utility of traditional chemical functional groups as used in chemical explanations. Our assessment employs a fundamental principle from computational learning theory: a good explanation of data should also compress the data. We introduce an unsupervised learning algorithm based on the Minimum Message Length (MML) principle that searches for substructures that compress around three million biologically relevant molecules. We demonstrate that the discovered substructures contain most human-curated functional groups as well as novel larger patterns with more specific functions. We also run our algorithm on 24 specific bioactivity prediction datasets to discover dataset-specific functional groups. Fingerprints constructed from dataset-specific functional groups are shown to significantly outperform other fingerprint representations, including the MACCS and Morgan fingerprint, when training ridge regression models on bioactivity regression tasks.
Learning Logical Rules using Minimum Message Length
Aug 08, 2025
Unifying probabilistic and logical learning is a key challenge in AI. We introduce a Bayesian inductive logic programming approach that learns minimum message length programs from noisy data. Our approach balances hypothesis complexity and data fit through priors, which explicitly favour more general programs, and a likelihood that favours accurate programs. Our experiments on several domains, including game playing and drug design, show that our method significantly outperforms previous methods, notably those that learn minimum description length programs. Our results also show that our approach is data-efficient and insensitive to example balance, including the ability to learn from exclusively positive examples.
Personalised Medicine: Establishing predictive machine learning models for drug responses in patient derived cell culture
Aug 23, 2024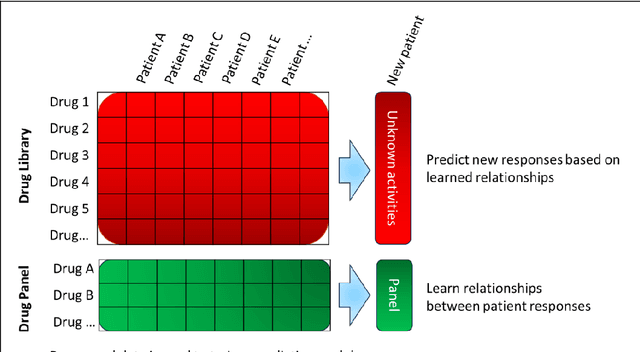
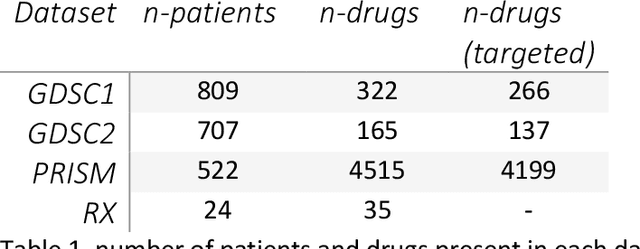

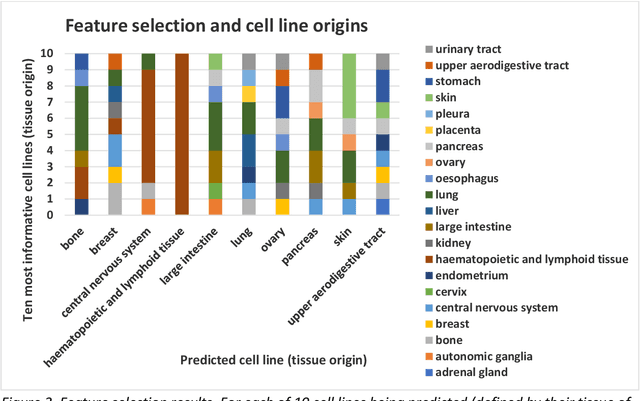
The concept of personalised medicine in cancer therapy is becoming increasingly important. There already exist drugs administered specifically for patients with tumours presenting well-defined mutations. However, the field is still in its infancy, and personalised treatments are far from being standard of care. Personalised medicine is often associated with the utilisation of omics data. Yet, implementation of multi-omics data has proven difficult, due to the variety and scale of the information within the data, as well as the complexity behind the myriad of interactions taking place within the cell. An alternative approach to precision medicine is to employ a function-based profile of the cell. This involves screening a range of drugs against patient derived cells. Here we demonstrate a proof-of-concept, where a collection of drug screens against a highly diverse set of patient-derived cell lines, are leveraged to identify putative treatment options for a 'new patient'. We show that this methodology is highly efficient in ranking the drugs according to their activity towards the target cells. We argue that this approach offers great potential, as activities can be efficiently imputed from various subsets of the drug treated cell lines that do not necessarily originate from the same tissue type.
Genesis: Towards the Automation of Systems Biology Research
Aug 20, 2024The cutting edge of applying AI to science is the closed-loop automation of scientific research: robot scientists. We have previously developed two robot scientists: `Adam' (for yeast functional biology), and `Eve' (for early-stage drug design)). We are now developing a next generation robot scientist Genesis. With Genesis we aim to demonstrate that an area of science can be investigated using robot scientists unambiguously faster, and at lower cost, than with human scientists. Here we report progress on the Genesis project. Genesis is designed to automatically improve system biology models with thousands of interacting causal components. When complete Genesis will be able to initiate and execute in parallel one thousand hypothesis-led closed-loop cycles of experiment per-day. Here we describe the core Genesis hardware: the one thousand computer-controlled $\mu$-bioreactors. For the integrated Mass Spectrometry platform we have developed AutonoMS, a system to automatically run, process, and analyse high-throughput experiments. We have also developed Genesis-DB, a database system designed to enable software agents access to large quantities of structured domain information. We have developed RIMBO (Revisions for Improvements of Models in Biology Ontology) to describe the planned hundreds of thousands of changes to the models. We have demonstrated the utility of this infrastructure by developed two relational learning bioinformatic projects. Finally, we describe LGEM+ a relational learning system for the automated abductive improvement of genome-scale metabolic models.
The Use of AI-Robotic Systems for Scientific Discovery
Jun 25, 2024

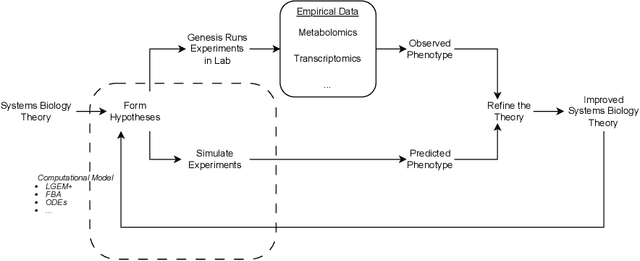
The process of developing theories and models and testing them with experiments is fundamental to the scientific method. Automating the entire scientific method then requires not only automation of the induction of theories from data, but also experimentation from design to implementation. This is the idea behind a robot scientist -- a coupled system of AI and laboratory robotics that has agency to test hypotheses with real-world experiments. In this chapter we explore some of the fundamentals of robot scientists in the philosophy of science. We also map the activities of a robot scientist to machine learning paradigms, and argue that the scientific method shares an analogy with active learning. We demonstrate these concepts using examples from previous robot scientists, and also from Genesis: a next generation robot scientist designed for research in systems biology, comprising a micro-fluidic system with 1000 computer-controlled micro-bioreactors and interpretable models based in controlled vocabularies and logic.
Scientific Hypothesis Generation by a Large Language Model: Laboratory Validation in Breast Cancer Treatment
May 20, 2024Large language models (LLMs) have transformed AI and achieved breakthrough performance on a wide range of tasks that require human intelligence. In science, perhaps the most interesting application of LLMs is for hypothesis formation. A feature of LLMs, which results from their probabilistic structure, is that the output text is not necessarily a valid inference from the training text. These are 'hallucinations', and are a serious problem in many applications. However, in science, hallucinations may be useful: they are novel hypotheses whose validity may be tested by laboratory experiments. Here we experimentally test the use of LLMs as a source of scientific hypotheses using the domain of breast cancer treatment. We applied the LLM GPT4 to hypothesize novel pairs of FDA-approved non-cancer drugs that target the MCF7 breast cancer cell line relative to the non-tumorigenic breast cell line MCF10A. In the first round of laboratory experiments GPT4 succeeded in discovering three drug combinations (out of 12 tested) with synergy scores above the positive controls. These combinations were itraconazole + atenolol, disulfiram + simvastatin and dipyridamole + mebendazole. GPT4 was then asked to generate new combinations after considering its initial results. It then discovered three more combinations with positive synergy scores (out of four tested), these were disulfiram + fulvestrant, mebendazole + quinacrine and disulfiram + quinacrine. A limitation of GPT4 as a generator of hypotheses was that its explanations for them were formulaic and unconvincing. We conclude that LLMs are an exciting novel source of scientific hypotheses.
Beating the Best: Improving on AlphaFold2 at Protein Structure Prediction
Jan 23, 2023
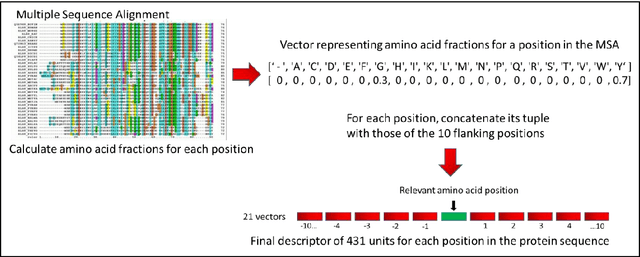
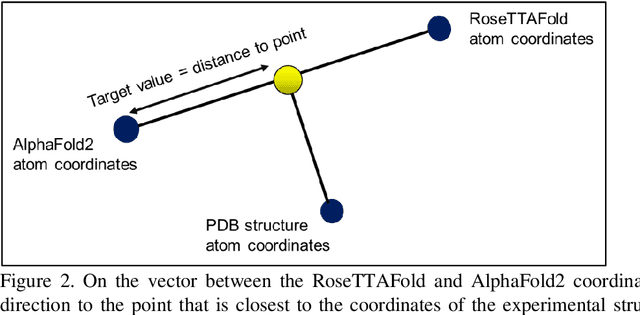
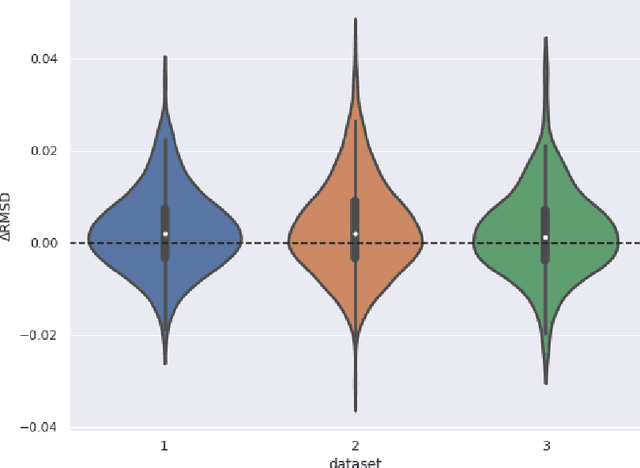
The goal of Protein Structure Prediction (PSP) problem is to predict a protein's 3D structure (confirmation) from its amino acid sequence. The problem has been a 'holy grail' of science since the Noble prize-winning work of Anfinsen demonstrated that protein conformation was determined by sequence. A recent and important step towards this goal was the development of AlphaFold2, currently the best PSP method. AlphaFold2 is probably the highest profile application of AI to science. Both AlphaFold2 and RoseTTAFold (another impressive PSP method) have been published and placed in the public domain (code & models). Stacking is a form of ensemble machine learning ML in which multiple baseline models are first learnt, then a meta-model is learnt using the outputs of the baseline level model to form a model that outperforms the base models. Stacking has been successful in many applications. We developed the ARStack PSP method by stacking AlphaFold2 and RoseTTAFold. ARStack significantly outperforms AlphaFold2. We rigorously demonstrate this using two sets of non-homologous proteins, and a test set of protein structures published after that of AlphaFold2 and RoseTTAFold. As more high quality prediction methods are published it is likely that ensemble methods will increasingly outperform any single method.
Transformative Machine Learning
Nov 08, 2018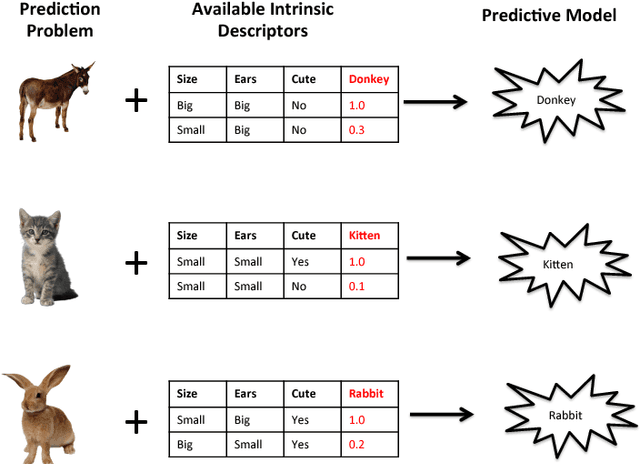
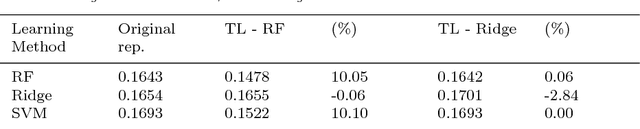
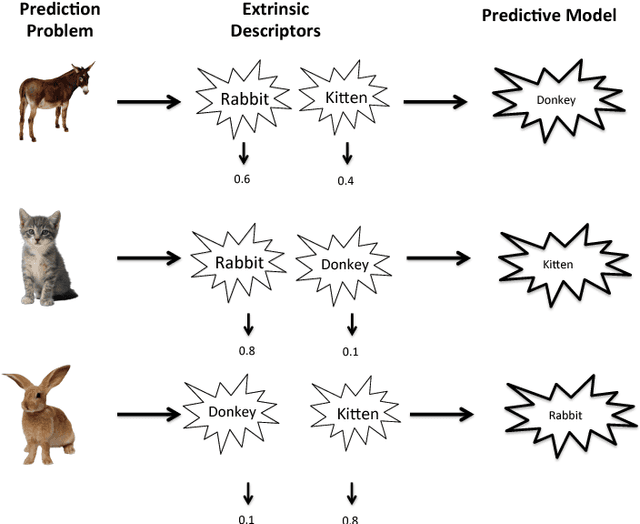
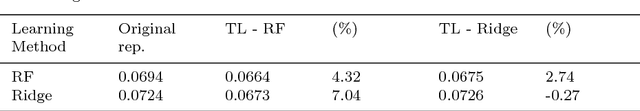
The key to success in machine learning (ML) is the use of effective data representations. Traditionally, data representations were hand-crafted. Recently it has been demonstrated that, given sufficient data, deep neural networks can learn effective implicit representations from simple input representations. However, for most scientific problems, the use of deep learning is not appropriate as the amount of available data is limited, and/or the output models must be explainable. Nevertheless, many scientific problems do have significant amounts of data available on related tasks, which makes them amenable to multi-task learning, i.e. learning many related problems simultaneously. Here we propose a novel and general representation learning approach for multi-task learning that works successfully with small amounts of data. The fundamental new idea is to transform an input intrinsic data representation (i.e., handcrafted features), to an extrinsic representation based on what a pre-trained set of models predict about the examples. This transformation has the dual advantages of producing significantly more accurate predictions, and providing explainable models. To demonstrate the utility of this transformative learning approach, we have applied it to three real-world scientific problems: drug-design (quantitative structure activity relationship learning), predicting human gene expression (across different tissue types and drug treatments), and meta-learning for machine learning (predicting which machine learning methods work best for a given problem). In all three problems, transformative machine learning significantly outperforms the best intrinsic representation.
Meta-QSAR: a large-scale application of meta-learning to drug design and discovery
Sep 12, 2017


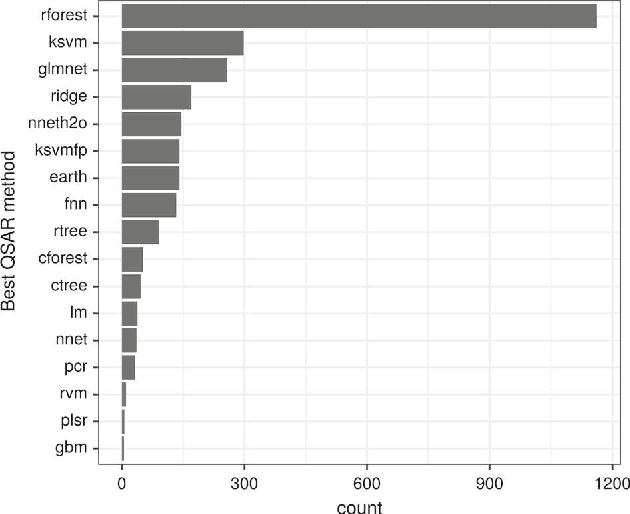
We investigate the learning of quantitative structure activity relationships (QSARs) as a case-study of meta-learning. This application area is of the highest societal importance, as it is a key step in the development of new medicines. The standard QSAR learning problem is: given a target (usually a protein) and a set of chemical compounds (small molecules) with associated bioactivities (e.g. inhibition of the target), learn a predictive mapping from molecular representation to activity. Although almost every type of machine learning method has been applied to QSAR learning there is no agreed single best way of learning QSARs, and therefore the problem area is well-suited to meta-learning. We first carried out the most comprehensive ever comparison of machine learning methods for QSAR learning: 18 regression methods, 6 molecular representations, applied to more than 2,700 QSAR problems. (These results have been made publicly available on OpenML and represent a valuable resource for testing novel meta-learning methods.) We then investigated the utility of algorithm selection for QSAR problems. We found that this meta-learning approach outperformed the best individual QSAR learning method (random forests using a molecular fingerprint representation) by up to 13%, on average. We conclude that meta-learning outperforms base-learning methods for QSAR learning, and as this investigation is one of the most extensive ever comparisons of base and meta-learning methods ever made, it provides evidence for the general effectiveness of meta-learning over base-learning.