 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMeta-QSAR: a large-scale application of meta-learning to drug design and discovery
Paper and Code
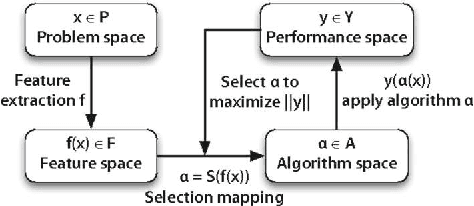


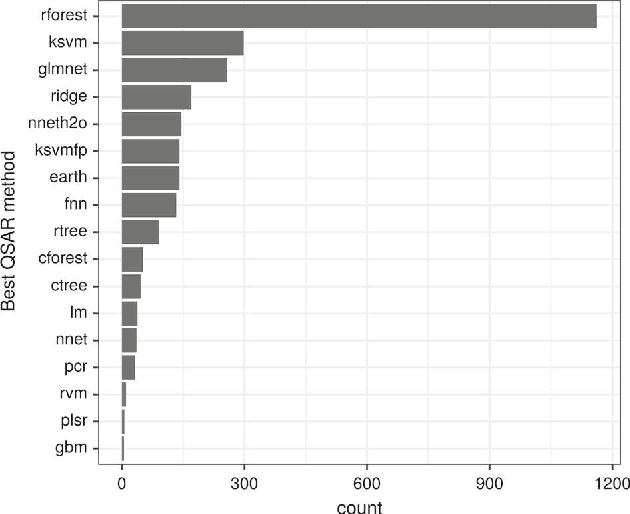
We investigate the learning of quantitative structure activity relationships (QSARs) as a case-study of meta-learning. This application area is of the highest societal importance, as it is a key step in the development of new medicines. The standard QSAR learning problem is: given a target (usually a protein) and a set of chemical compounds (small molecules) with associated bioactivities (e.g. inhibition of the target), learn a predictive mapping from molecular representation to activity. Although almost every type of machine learning method has been applied to QSAR learning there is no agreed single best way of learning QSARs, and therefore the problem area is well-suited to meta-learning. We first carried out the most comprehensive ever comparison of machine learning methods for QSAR learning: 18 regression methods, 6 molecular representations, applied to more than 2,700 QSAR problems. (These results have been made publicly available on OpenML and represent a valuable resource for testing novel meta-learning methods.) We then investigated the utility of algorithm selection for QSAR problems. We found that this meta-learning approach outperformed the best individual QSAR learning method (random forests using a molecular fingerprint representation) by up to 13%, on average. We conclude that meta-learning outperforms base-learning methods for QSAR learning, and as this investigation is one of the most extensive ever comparisons of base and meta-learning methods ever made, it provides evidence for the general effectiveness of meta-learning over base-learning.