 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePersonalised Medicine: Establishing predictive machine learning models for drug responses in patient derived cell culture
Aug 23, 2024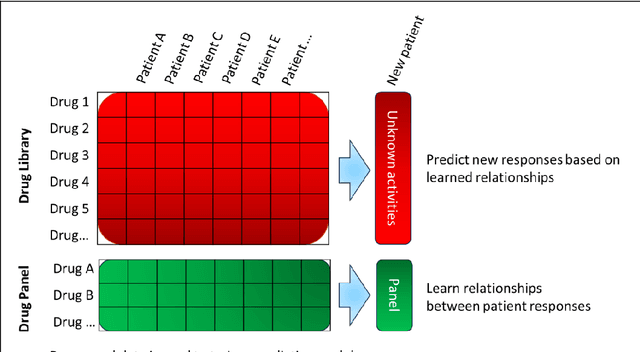
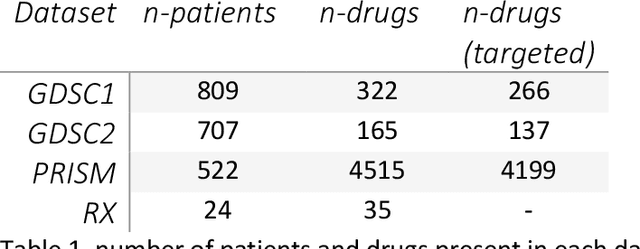

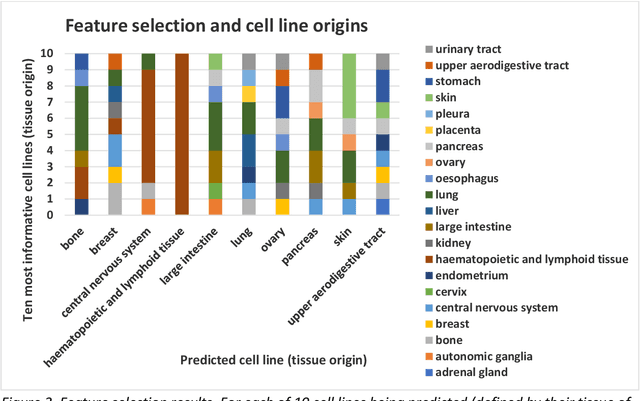
The concept of personalised medicine in cancer therapy is becoming increasingly important. There already exist drugs administered specifically for patients with tumours presenting well-defined mutations. However, the field is still in its infancy, and personalised treatments are far from being standard of care. Personalised medicine is often associated with the utilisation of omics data. Yet, implementation of multi-omics data has proven difficult, due to the variety and scale of the information within the data, as well as the complexity behind the myriad of interactions taking place within the cell. An alternative approach to precision medicine is to employ a function-based profile of the cell. This involves screening a range of drugs against patient derived cells. Here we demonstrate a proof-of-concept, where a collection of drug screens against a highly diverse set of patient-derived cell lines, are leveraged to identify putative treatment options for a 'new patient'. We show that this methodology is highly efficient in ranking the drugs according to their activity towards the target cells. We argue that this approach offers great potential, as activities can be efficiently imputed from various subsets of the drug treated cell lines that do not necessarily originate from the same tissue type.
The Future of Fundamental Science Led by Generative Closed-Loop Artificial Intelligence
Jul 09, 2023



Recent advances in machine learning and AI, including Generative AI and LLMs, are disrupting technological innovation, product development, and society as a whole. AI's contribution to technology can come from multiple approaches that require access to large training data sets and clear performance evaluation criteria, ranging from pattern recognition and classification to generative models. Yet, AI has contributed less to fundamental science in part because large data sets of high-quality data for scientific practice and model discovery are more difficult to access. Generative AI, in general, and Large Language Models in particular, may represent an opportunity to augment and accelerate the scientific discovery of fundamental deep science with quantitative models. Here we explore and investigate aspects of an AI-driven, automated, closed-loop approach to scientific discovery, including self-driven hypothesis generation and open-ended autonomous exploration of the hypothesis space. Integrating AI-driven automation into the practice of science would mitigate current problems, including the replication of findings, systematic production of data, and ultimately democratisation of the scientific process. Realising these possibilities requires a vision for augmented AI coupled with a diversity of AI approaches able to deal with fundamental aspects of causality analysis and model discovery while enabling unbiased search across the space of putative explanations. These advances hold the promise to unleash AI's potential for searching and discovering the fundamental structure of our world beyond what human scientists have been able to achieve. Such a vision would push the boundaries of new fundamental science rather than automatize current workflows and instead open doors for technological innovation to tackle some of the greatest challenges facing humanity today.
ML-Schema: Exposing the Semantics of Machine Learning with Schemas and Ontologies
Jul 14, 2018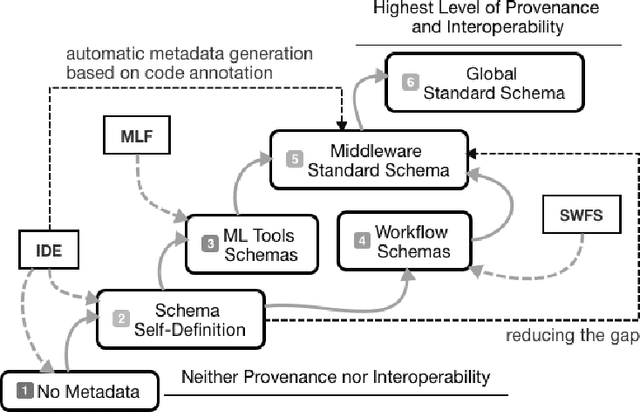
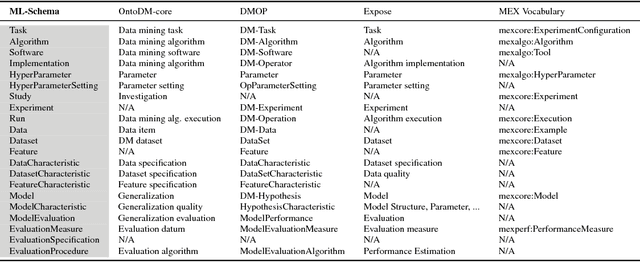
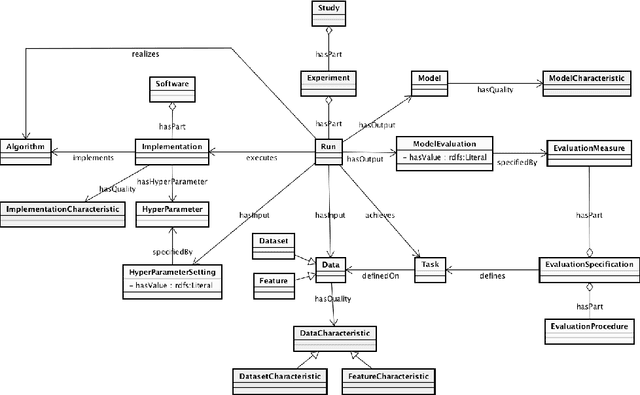
The ML-Schema, proposed by the W3C Machine Learning Schema Community Group, is a top-level ontology that provides a set of classes, properties, and restrictions for representing and interchanging information on machine learning algorithms, datasets, and experiments. It can be easily extended and specialized and it is also mapped to other more domain-specific ontologies developed in the area of machine learning and data mining. In this paper we overview existing state-of-the-art machine learning interchange formats and present the first release of ML-Schema, a canonical format resulted of more than seven years of experience among different research institutions. We argue that exposing semantics of machine learning algorithms, models, and experiments through a canonical format may pave the way to better interpretability and to realistically achieve the full interoperability of experiments regardless of platform or adopted workflow solution.
Meta-QSAR: a large-scale application of meta-learning to drug design and discovery
Sep 12, 2017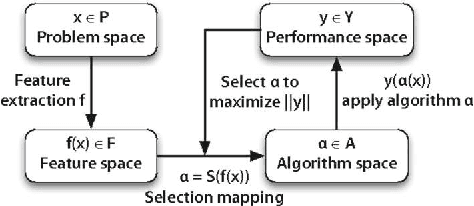


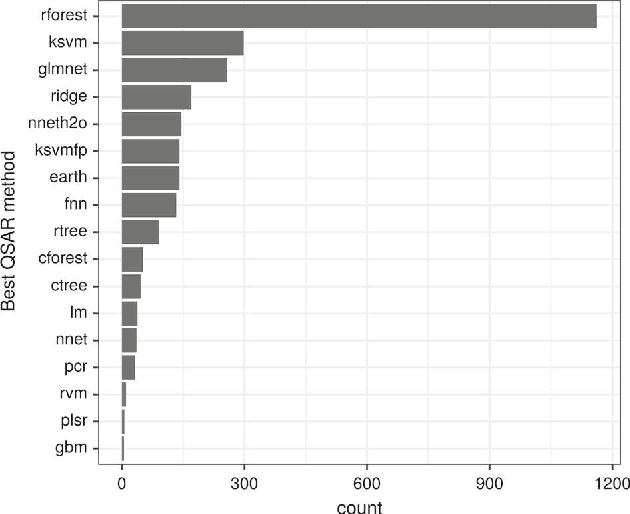
We investigate the learning of quantitative structure activity relationships (QSARs) as a case-study of meta-learning. This application area is of the highest societal importance, as it is a key step in the development of new medicines. The standard QSAR learning problem is: given a target (usually a protein) and a set of chemical compounds (small molecules) with associated bioactivities (e.g. inhibition of the target), learn a predictive mapping from molecular representation to activity. Although almost every type of machine learning method has been applied to QSAR learning there is no agreed single best way of learning QSARs, and therefore the problem area is well-suited to meta-learning. We first carried out the most comprehensive ever comparison of machine learning methods for QSAR learning: 18 regression methods, 6 molecular representations, applied to more than 2,700 QSAR problems. (These results have been made publicly available on OpenML and represent a valuable resource for testing novel meta-learning methods.) We then investigated the utility of algorithm selection for QSAR problems. We found that this meta-learning approach outperformed the best individual QSAR learning method (random forests using a molecular fingerprint representation) by up to 13%, on average. We conclude that meta-learning outperforms base-learning methods for QSAR learning, and as this investigation is one of the most extensive ever comparisons of base and meta-learning methods ever made, it provides evidence for the general effectiveness of meta-learning over base-learning.