 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePersonalised Medicine: Establishing predictive machine learning models for drug responses in patient derived cell culture
Aug 23, 2024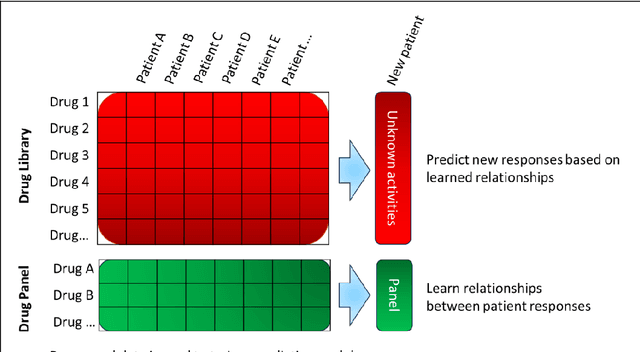
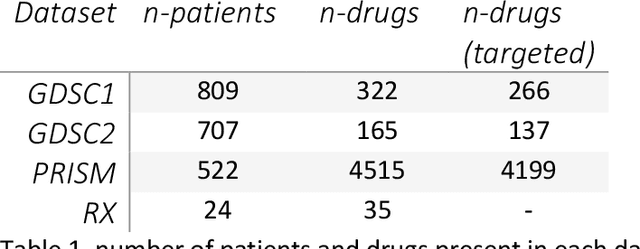

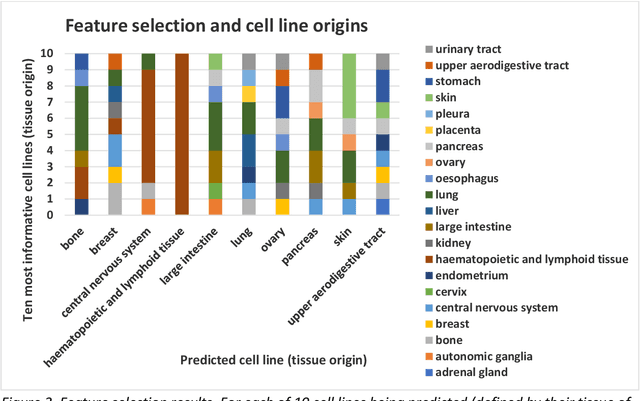
The concept of personalised medicine in cancer therapy is becoming increasingly important. There already exist drugs administered specifically for patients with tumours presenting well-defined mutations. However, the field is still in its infancy, and personalised treatments are far from being standard of care. Personalised medicine is often associated with the utilisation of omics data. Yet, implementation of multi-omics data has proven difficult, due to the variety and scale of the information within the data, as well as the complexity behind the myriad of interactions taking place within the cell. An alternative approach to precision medicine is to employ a function-based profile of the cell. This involves screening a range of drugs against patient derived cells. Here we demonstrate a proof-of-concept, where a collection of drug screens against a highly diverse set of patient-derived cell lines, are leveraged to identify putative treatment options for a 'new patient'. We show that this methodology is highly efficient in ranking the drugs according to their activity towards the target cells. We argue that this approach offers great potential, as activities can be efficiently imputed from various subsets of the drug treated cell lines that do not necessarily originate from the same tissue type.
Scientific Hypothesis Generation by a Large Language Model: Laboratory Validation in Breast Cancer Treatment
May 20, 2024Large language models (LLMs) have transformed AI and achieved breakthrough performance on a wide range of tasks that require human intelligence. In science, perhaps the most interesting application of LLMs is for hypothesis formation. A feature of LLMs, which results from their probabilistic structure, is that the output text is not necessarily a valid inference from the training text. These are 'hallucinations', and are a serious problem in many applications. However, in science, hallucinations may be useful: they are novel hypotheses whose validity may be tested by laboratory experiments. Here we experimentally test the use of LLMs as a source of scientific hypotheses using the domain of breast cancer treatment. We applied the LLM GPT4 to hypothesize novel pairs of FDA-approved non-cancer drugs that target the MCF7 breast cancer cell line relative to the non-tumorigenic breast cell line MCF10A. In the first round of laboratory experiments GPT4 succeeded in discovering three drug combinations (out of 12 tested) with synergy scores above the positive controls. These combinations were itraconazole + atenolol, disulfiram + simvastatin and dipyridamole + mebendazole. GPT4 was then asked to generate new combinations after considering its initial results. It then discovered three more combinations with positive synergy scores (out of four tested), these were disulfiram + fulvestrant, mebendazole + quinacrine and disulfiram + quinacrine. A limitation of GPT4 as a generator of hypotheses was that its explanations for them were formulaic and unconvincing. We conclude that LLMs are an exciting novel source of scientific hypotheses.
Beating the Best: Improving on AlphaFold2 at Protein Structure Prediction
Jan 23, 2023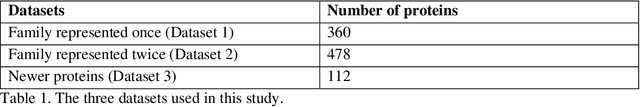
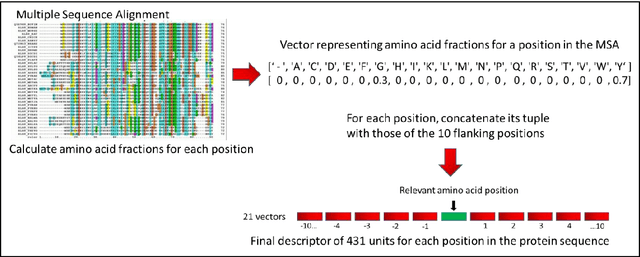
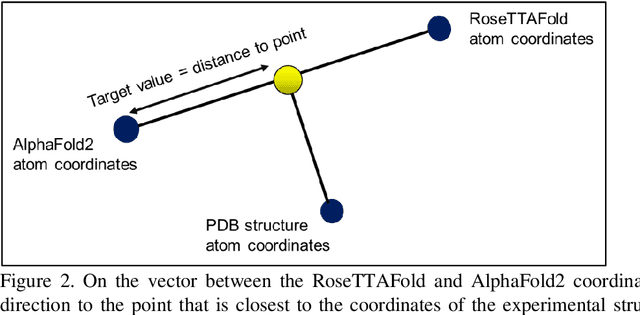
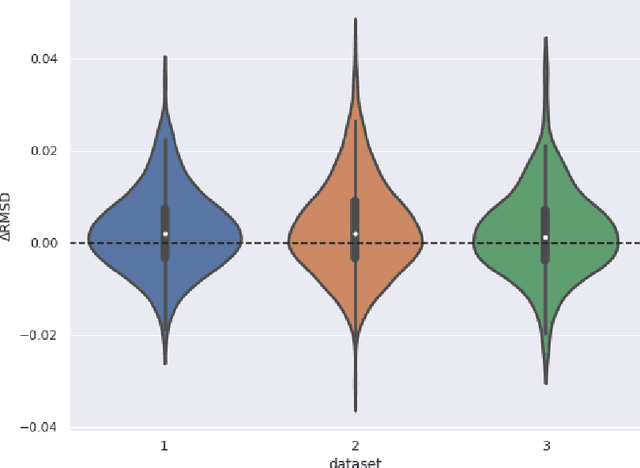
The goal of Protein Structure Prediction (PSP) problem is to predict a protein's 3D structure (confirmation) from its amino acid sequence. The problem has been a 'holy grail' of science since the Noble prize-winning work of Anfinsen demonstrated that protein conformation was determined by sequence. A recent and important step towards this goal was the development of AlphaFold2, currently the best PSP method. AlphaFold2 is probably the highest profile application of AI to science. Both AlphaFold2 and RoseTTAFold (another impressive PSP method) have been published and placed in the public domain (code & models). Stacking is a form of ensemble machine learning ML in which multiple baseline models are first learnt, then a meta-model is learnt using the outputs of the baseline level model to form a model that outperforms the base models. Stacking has been successful in many applications. We developed the ARStack PSP method by stacking AlphaFold2 and RoseTTAFold. ARStack significantly outperforms AlphaFold2. We rigorously demonstrate this using two sets of non-homologous proteins, and a test set of protein structures published after that of AlphaFold2 and RoseTTAFold. As more high quality prediction methods are published it is likely that ensemble methods will increasingly outperform any single method.