 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnhancing Ligand Pose Sampling for Molecular Docking
Nov 30, 2023Deep learning promises to dramatically improve scoring functions for molecular docking, leading to substantial advances in binding pose prediction and virtual screening. To train scoring functions-and to perform molecular docking-one must generate a set of candidate ligand binding poses. Unfortunately, the sampling protocols currently used to generate candidate poses frequently fail to produce any poses close to the correct, experimentally determined pose, unless information about the correct pose is provided. This limits the accuracy of learned scoring functions and molecular docking. Here, we describe two improved protocols for pose sampling: GLOW (auGmented sampLing with sOftened vdW potential) and a novel technique named IVES (IteratiVe Ensemble Sampling). Our benchmarking results demonstrate the effectiveness of our methods in improving the likelihood of sampling accurate poses, especially for binding pockets whose shape changes substantially when different ligands bind. This improvement is observed across both experimentally determined and AlphaFold-generated protein structures. Additionally, we present datasets of candidate ligand poses generated using our methods for each of around 5,000 protein-ligand cross-docking pairs, for training and testing scoring functions. To benefit the research community, we provide these cross-docking datasets and an open-source Python implementation of GLOW and IVES at https://github.com/drorlab/GLOW_IVES .
FlexVDW: A machine learning approach to account for protein flexibility in ligand docking
Mar 20, 2023Most widely used ligand docking methods assume a rigid protein structure. This leads to problems when the structure of the target protein deforms upon ligand binding. In particular, the ligand's true binding pose is often scored very unfavorably due to apparent clashes between ligand and protein atoms, which lead to extremely high values of the calculated van der Waals energy term. Traditionally, this problem has been addressed by explicitly searching for receptor conformations to account for the flexibility of the receptor in ligand binding. Here we present a deep learning model trained to take receptor flexibility into account implicitly when predicting van der Waals energy. We show that incorporating this machine-learned energy term into a state-of-the-art physics-based scoring function improves small molecule ligand pose prediction results in cases with substantial protein deformation, without degrading performance in cases with minimal protein deformation. This work demonstrates the feasibility of learning effects of protein flexibility on ligand binding without explicitly modeling changes in protein structure.
ATOM3D: Tasks On Molecules in Three Dimensions
Dec 07, 2020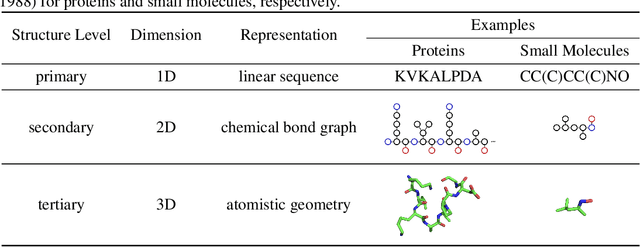
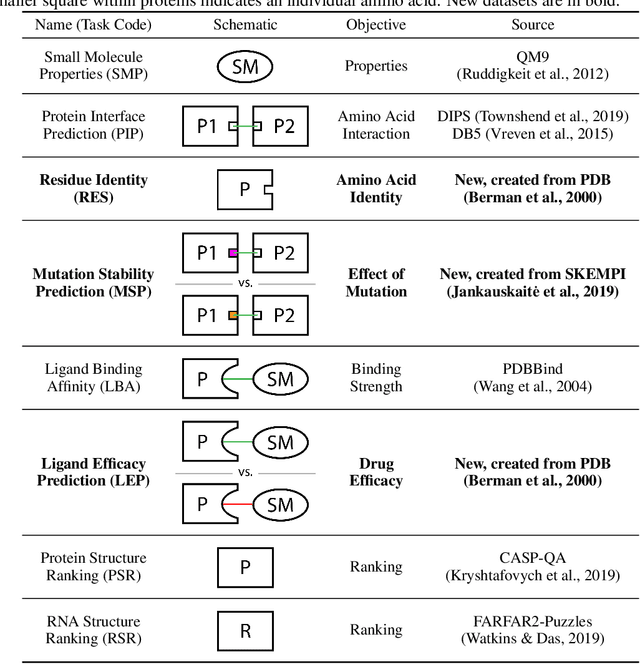
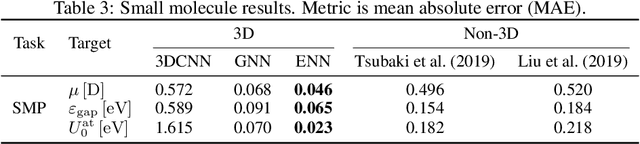
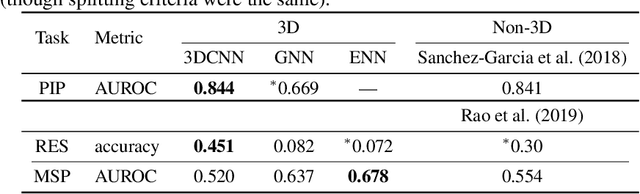
Computational methods that operate directly on three-dimensional molecular structure hold large potential to solve important questions in biology and chemistry. In particular deep neural networks have recently gained significant attention. In this work we present ATOM3D, a collection of both novel and existing datasets spanning several key classes of biomolecules, to systematically assess such learning methods. We develop three-dimensional molecular learning networks for each of these tasks, finding that they consistently improve performance relative to one- and two-dimensional methods. The specific choice of architecture proves to be critical for performance, with three-dimensional convolutional networks excelling at tasks involving complex geometries, while graph networks perform well on systems requiring detailed positional information. Furthermore, equivariant networks show significant promise. Our results indicate many molecular problems stand to gain from three-dimensional molecular learning. All code and datasets can be accessed via https://www.atom3d.ai .
Protein model quality assessment using rotation-equivariant, hierarchical neural networks
Nov 27, 2020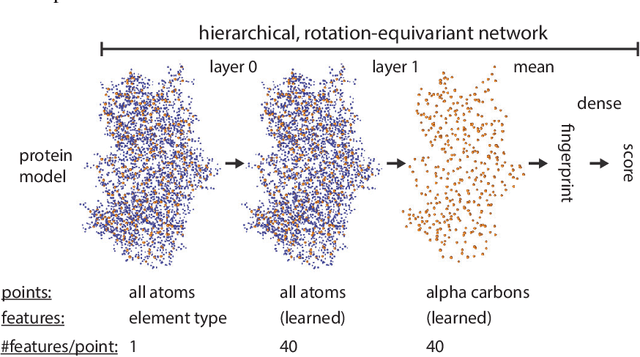
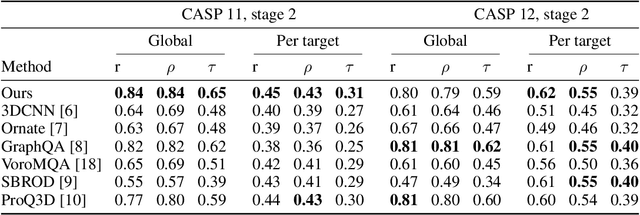
Proteins are miniature machines whose function depends on their three-dimensional (3D) structure. Determining this structure computationally remains an unsolved grand challenge. A major bottleneck involves selecting the most accurate structural model among a large pool of candidates, a task addressed in model quality assessment. Here, we present a novel deep learning approach to assess the quality of a protein model. Our network builds on a point-based representation of the atomic structure and rotation-equivariant convolutions at different levels of structural resolution. These combined aspects allow the network to learn end-to-end from entire protein structures. Our method achieves state-of-the-art results in scoring protein models submitted to recent rounds of CASP, a blind prediction community experiment. Particularly striking is that our method does not use physics-inspired energy terms and does not rely on the availability of additional information (beyond the atomic structure of the individual protein model), such as sequence alignments of multiple proteins.
Learning from Protein Structure with Geometric Vector Perceptrons
Sep 03, 2020
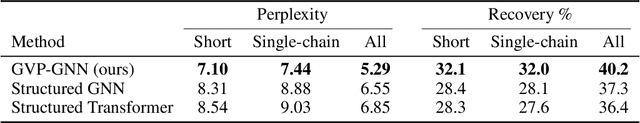
Learning on 3D structures of large biomolecules is emerging as a distinct area in machine learning, but there has yet to emerge a unifying network architecture that simultaneously leverages the graph-structured and geometric aspects of the problem domain. To address this gap, we introduce geometric vector perceptrons, which extend standard dense layers to operate on collections of Euclidean vectors. Graph neural networks equipped with such layers are able to perform both geometric and relational reasoning on efficient and natural representations of macromolecular structure. We demonstrate our approach on two important problems in learning from protein structure: model quality assessment and computational protein design. Our approach improves over existing classes of architectures, including state-of-the-art graph-based and voxel-based methods.
Tiramisu: A Code Optimization Framework for High Performance Systems
Sep 26, 2018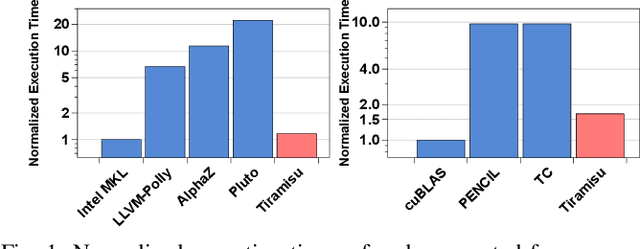


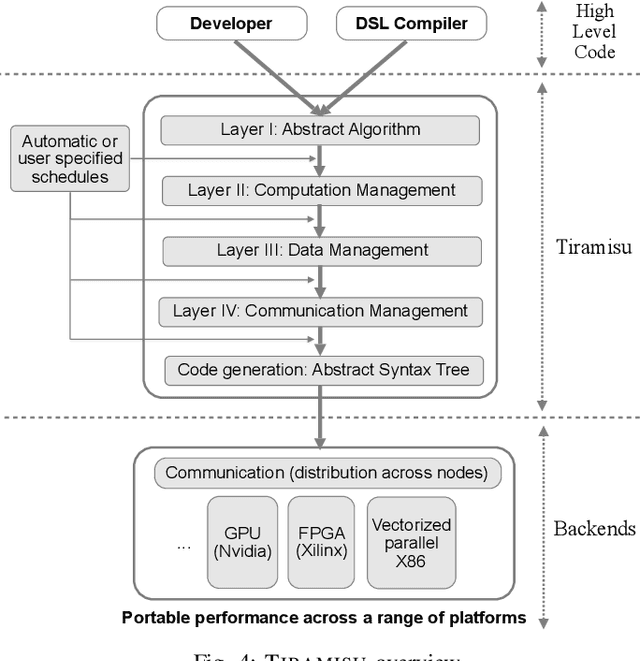
This paper introduces Tiramisu, a polyhedral framework designed to generate high performance code for multiple platforms including multicores, GPUs, and distributed machines. Tiramisu introduces a scheduling language with novel extensions to explicitly manage the complexities that arise when targeting these systems. The extensions include explicit communication, synchronization, and mapping buffers to different memory hierarchies. Tiramisu relies on a flexible representation based on the polyhedral model and explicitly uses a well-defined four-level IR that allows full separation between the algorithms, loop transformations, data-layouts, and communication. This separation simplifies targeting multiple hardware architectures with the same algorithm. We evaluate Tiramisu by writing a set of image processing and stencil benchmarks and compare it with state-of-the-art compilers. We show that Tiramisu matches or outperforms existing compilers on different hardware architectures, including multicore CPUs, GPUs, and distributed machines.