 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeometric Latent Diffusion Models for 3D Molecule Generation
May 02, 2023



Generative models, especially diffusion models (DMs), have achieved promising results for generating feature-rich geometries and advancing foundational science problems such as molecule design. Inspired by the recent huge success of Stable (latent) Diffusion models, we propose a novel and principled method for 3D molecule generation named Geometric Latent Diffusion Models (GeoLDM). GeoLDM is the first latent DM model for the molecular geometry domain, composed of autoencoders encoding structures into continuous latent codes and DMs operating in the latent space. Our key innovation is that for modeling the 3D molecular geometries, we capture its critical roto-translational equivariance constraints by building a point-structured latent space with both invariant scalars and equivariant tensors. Extensive experiments demonstrate that GeoLDM can consistently achieve better performance on multiple molecule generation benchmarks, with up to 7\% improvement for the valid percentage of large biomolecules. Results also demonstrate GeoLDM's higher capacity for controllable generation thanks to the latent modeling. Code is provided at \url{https://github.com/MinkaiXu/GeoLDM}.
Harnessing Simulation for Molecular Embeddings
Feb 04, 2023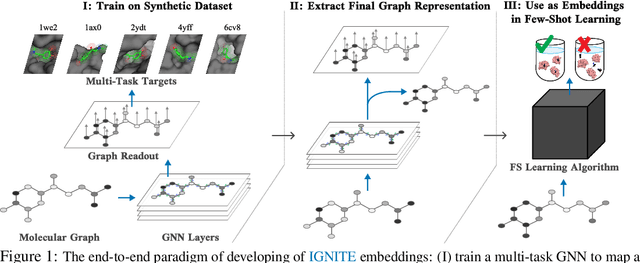

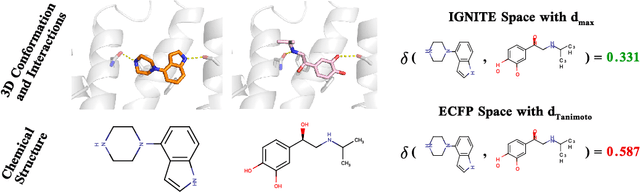
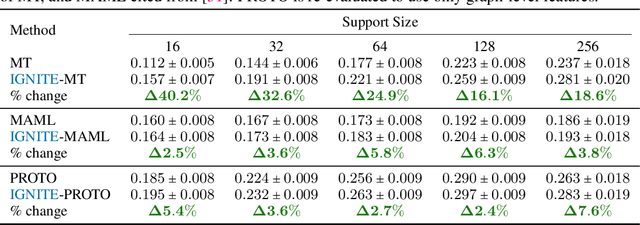
While deep learning has unlocked advances in computational biology once thought to be decades away, extending deep learning techniques to the molecular domain has proven challenging, as labeled data is scarce and the benefit from self-supervised learning can be negligible in many cases. In this work, we explore a different approach. Inspired by methods in deep reinforcement learning and robotics, we explore harnessing physics-based molecular simulation to develop molecular embeddings. By fitting a Graph Neural Network to simulation data, molecules that display similar interactions with biological targets under simulation develop similar representations in the embedding space. These embeddings can then be used to initialize the feature space of down-stream models trained on real-world data to encode information learned during simulation into a molecular prediction task. Our experimental findings indicate this approach improves the performance of existing deep learning models on real-world molecular prediction tasks by as much as 38% with minimal modification to the downstream model and no hyperparameter tuning.
Learning from Protein Structure with Geometric Vector Perceptrons
Sep 03, 2020
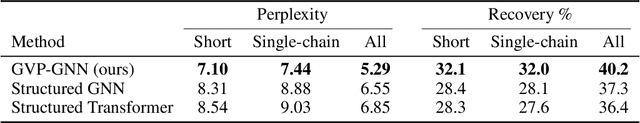
Learning on 3D structures of large biomolecules is emerging as a distinct area in machine learning, but there has yet to emerge a unifying network architecture that simultaneously leverages the graph-structured and geometric aspects of the problem domain. To address this gap, we introduce geometric vector perceptrons, which extend standard dense layers to operate on collections of Euclidean vectors. Graph neural networks equipped with such layers are able to perform both geometric and relational reasoning on efficient and natural representations of macromolecular structure. We demonstrate our approach on two important problems in learning from protein structure: model quality assessment and computational protein design. Our approach improves over existing classes of architectures, including state-of-the-art graph-based and voxel-based methods.
Hierarchical, rotation-equivariant neural networks to predict the structure of protein complexes
Jun 05, 2020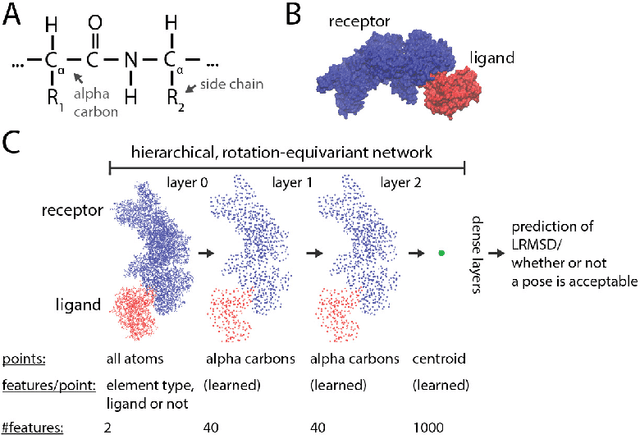
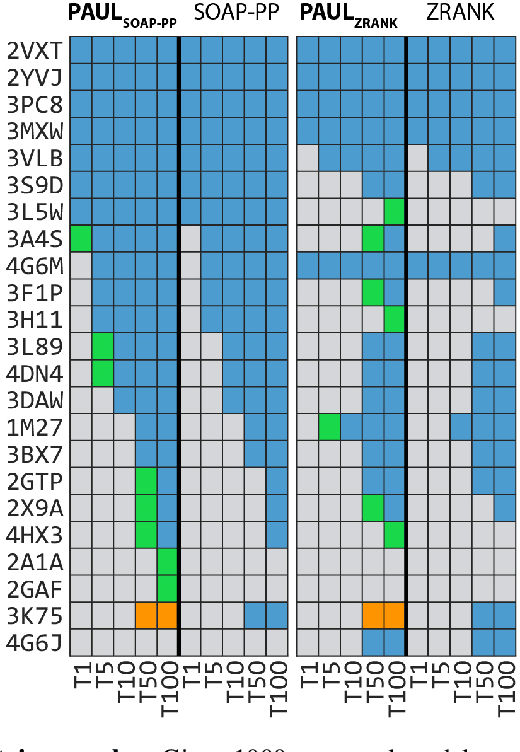
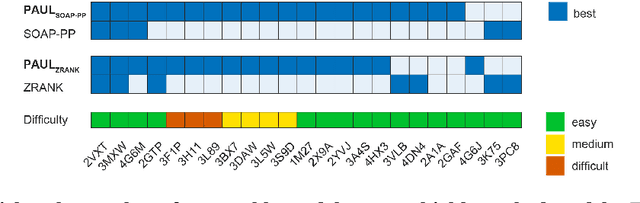

Predicting the structure of multi-protein complexes is a grand challenge in biochemistry, with major implications for basic science and drug discovery. Computational structure prediction methods generally leverage pre-defined structural features to distinguish accurate structural models from less accurate ones. This raises the question of whether it is possible to learn characteristics of accurate models directly from atomic coordinates of protein complexes, with no prior assumptions. Here we introduce a machine learning method that learns directly from the 3D positions of all atoms to identify accurate models of protein complexes, without using any pre-computed physics-inspired or statistical terms. Our neural network architecture combines multiple ingredients that together enable end-to-end learning from molecular structures containing tens of thousands of atoms: a point-based representation of atoms, equivariance with respect to rotation and translation, local convolutions, and hierarchical subsampling operations. When used in combination with previously developed scoring functions, our network substantially improves the identification of accurate structural models among a large set of possible models. Our network can also be used to predict the accuracy of a given structural model in absolute terms. The architecture we present is readily applicable to other tasks involving learning on 3D structures of large atomic systems.