 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHierarchical, rotation-equivariant neural networks to predict the structure of protein complexes
Paper and Code
Jun 05, 2020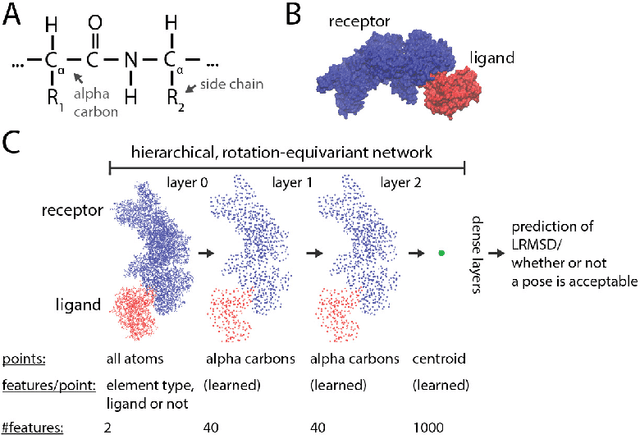
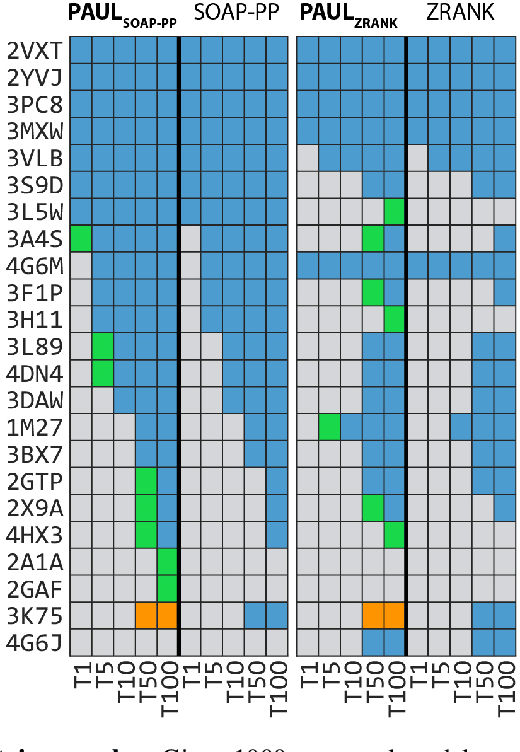
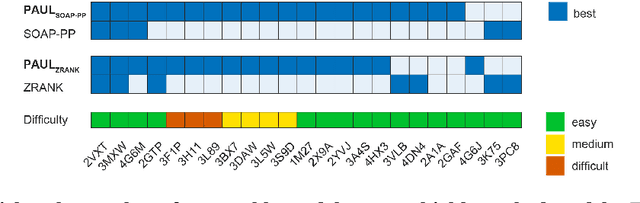

Predicting the structure of multi-protein complexes is a grand challenge in biochemistry, with major implications for basic science and drug discovery. Computational structure prediction methods generally leverage pre-defined structural features to distinguish accurate structural models from less accurate ones. This raises the question of whether it is possible to learn characteristics of accurate models directly from atomic coordinates of protein complexes, with no prior assumptions. Here we introduce a machine learning method that learns directly from the 3D positions of all atoms to identify accurate models of protein complexes, without using any pre-computed physics-inspired or statistical terms. Our neural network architecture combines multiple ingredients that together enable end-to-end learning from molecular structures containing tens of thousands of atoms: a point-based representation of atoms, equivariance with respect to rotation and translation, local convolutions, and hierarchical subsampling operations. When used in combination with previously developed scoring functions, our network substantially improves the identification of accurate structural models among a large set of possible models. Our network can also be used to predict the accuracy of a given structural model in absolute terms. The architecture we present is readily applicable to other tasks involving learning on 3D structures of large atomic systems.