 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFlexVDW: A machine learning approach to account for protein flexibility in ligand docking
Mar 20, 2023Most widely used ligand docking methods assume a rigid protein structure. This leads to problems when the structure of the target protein deforms upon ligand binding. In particular, the ligand's true binding pose is often scored very unfavorably due to apparent clashes between ligand and protein atoms, which lead to extremely high values of the calculated van der Waals energy term. Traditionally, this problem has been addressed by explicitly searching for receptor conformations to account for the flexibility of the receptor in ligand binding. Here we present a deep learning model trained to take receptor flexibility into account implicitly when predicting van der Waals energy. We show that incorporating this machine-learned energy term into a state-of-the-art physics-based scoring function improves small molecule ligand pose prediction results in cases with substantial protein deformation, without degrading performance in cases with minimal protein deformation. This work demonstrates the feasibility of learning effects of protein flexibility on ligand binding without explicitly modeling changes in protein structure.
Harnessing Simulation for Molecular Embeddings
Feb 04, 2023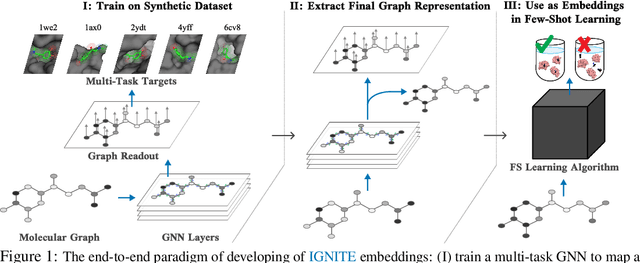

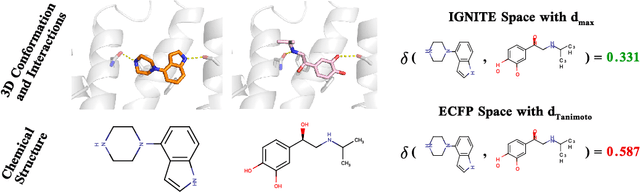
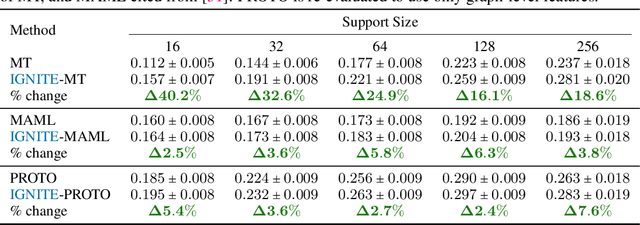
While deep learning has unlocked advances in computational biology once thought to be decades away, extending deep learning techniques to the molecular domain has proven challenging, as labeled data is scarce and the benefit from self-supervised learning can be negligible in many cases. In this work, we explore a different approach. Inspired by methods in deep reinforcement learning and robotics, we explore harnessing physics-based molecular simulation to develop molecular embeddings. By fitting a Graph Neural Network to simulation data, molecules that display similar interactions with biological targets under simulation develop similar representations in the embedding space. These embeddings can then be used to initialize the feature space of down-stream models trained on real-world data to encode information learned during simulation into a molecular prediction task. Our experimental findings indicate this approach improves the performance of existing deep learning models on real-world molecular prediction tasks by as much as 38% with minimal modification to the downstream model and no hyperparameter tuning.