 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAligning Target-Aware Molecule Diffusion Models with Exact Energy Optimization
Jul 01, 2024Generating ligand molecules for specific protein targets, known as structure-based drug design, is a fundamental problem in therapeutics development and biological discovery. Recently, target-aware generative models, especially diffusion models, have shown great promise in modeling protein-ligand interactions and generating candidate drugs. However, existing models primarily focus on learning the chemical distribution of all drug candidates, which lacks effective steerability on the chemical quality of model generations. In this paper, we propose a novel and general alignment framework to align pretrained target diffusion models with preferred functional properties, named AliDiff. AliDiff shifts the target-conditioned chemical distribution towards regions with higher binding affinity and structural rationality, specified by user-defined reward functions, via the preference optimization approach. To avoid the overfitting problem in common preference optimization objectives, we further develop an improved Exact Energy Preference Optimization method to yield an exact and efficient alignment of the diffusion models, and provide the closed-form expression for the converged distribution. Empirical studies on the CrossDocked2020 benchmark show that AliDiff can generate molecules with state-of-the-art binding energies with up to -7.07 Avg. Vina Score, while maintaining strong molecular properties.
Geometric Latent Diffusion Models for 3D Molecule Generation
May 02, 2023



Generative models, especially diffusion models (DMs), have achieved promising results for generating feature-rich geometries and advancing foundational science problems such as molecule design. Inspired by the recent huge success of Stable (latent) Diffusion models, we propose a novel and principled method for 3D molecule generation named Geometric Latent Diffusion Models (GeoLDM). GeoLDM is the first latent DM model for the molecular geometry domain, composed of autoencoders encoding structures into continuous latent codes and DMs operating in the latent space. Our key innovation is that for modeling the 3D molecular geometries, we capture its critical roto-translational equivariance constraints by building a point-structured latent space with both invariant scalars and equivariant tensors. Extensive experiments demonstrate that GeoLDM can consistently achieve better performance on multiple molecule generation benchmarks, with up to 7\% improvement for the valid percentage of large biomolecules. Results also demonstrate GeoLDM's higher capacity for controllable generation thanks to the latent modeling. Code is provided at \url{https://github.com/MinkaiXu/GeoLDM}.
ATOM3D: Tasks On Molecules in Three Dimensions
Dec 07, 2020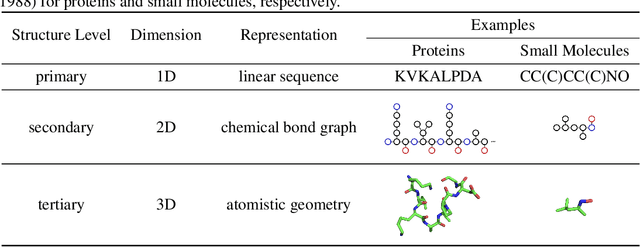
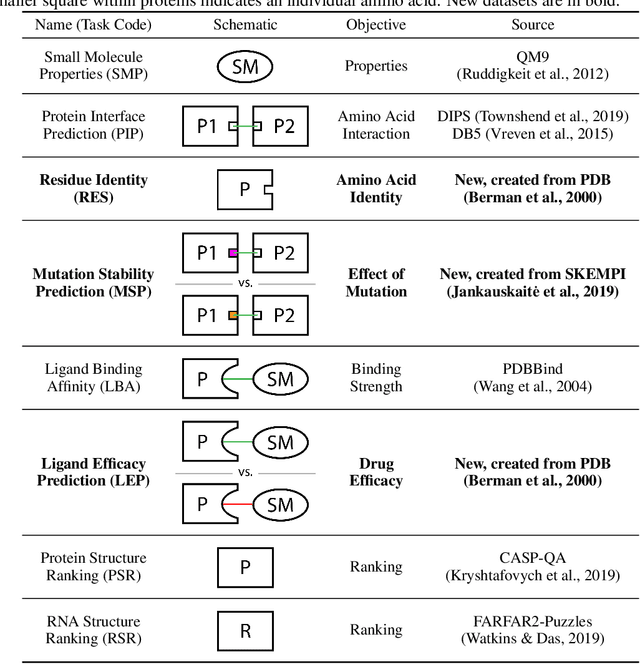
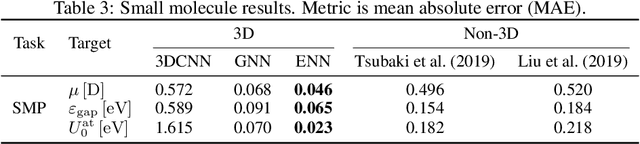
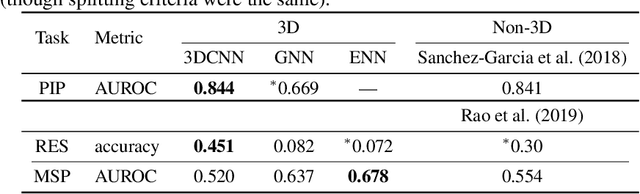
Computational methods that operate directly on three-dimensional molecular structure hold large potential to solve important questions in biology and chemistry. In particular deep neural networks have recently gained significant attention. In this work we present ATOM3D, a collection of both novel and existing datasets spanning several key classes of biomolecules, to systematically assess such learning methods. We develop three-dimensional molecular learning networks for each of these tasks, finding that they consistently improve performance relative to one- and two-dimensional methods. The specific choice of architecture proves to be critical for performance, with three-dimensional convolutional networks excelling at tasks involving complex geometries, while graph networks perform well on systems requiring detailed positional information. Furthermore, equivariant networks show significant promise. Our results indicate many molecular problems stand to gain from three-dimensional molecular learning. All code and datasets can be accessed via https://www.atom3d.ai .