 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutonomous FAIR Digital Objects: From Passive Assertions to Active Knowledge
May 11, 2026Scientific knowledge on the Web is published as passive assertions and cannot decide when to validate evidence, reconcile contradictions, or update confidence as findings accumulate. Curation depends on centralised middleware and institutional continuity, but when registries close, active stewardship stops even when data remain online. We advance the concept of Autonomous FAIR Digital Objects (aFDOs) from an abstract idea to an operational model, to offer a route from passive scientific publication toward accountable, standards-aligned automation that can outlive its publishing institutions. aFDO augments FDOs with three capabilities anchored in Semantic Web standards, namely 1) a policy layer over RDF-star aligned with PROV-O, SHACL, and ODRL for portable condition-action rules, 2) an announcement layer over ActivityStreams 2.0 that bounds per-announcement evaluation cost, and 3) an agreement layer that resolves multi-source contradictions through reputation and confidence weighted agreement under a bounded adversarial model. We provide a formal definition that distinguishes policy specifications, event handlers, and communication interfaces. We evaluate an open reference implementation on 4,305 FDOs grounded in rare-disease ontologies, namely ClinVar, HPO, and Orphanet, combined with controlled synthetic observations. The consensus mechanism resolves 56.3% of 3,914 naturally occurring ClinVar conflicts where multiple submitters disagree and an expert panel has subsequently adjudicated. Under Sybil, collusion, and poisoning attacks, the mechanism degrades gracefully within its design Byzantine-tolerance bound (f < n/5), and fails as predicted beyond that bound.
From Large Language Models to Knowledge Graphs for Biomarker Discovery in Cancer
Oct 12, 2023



Domain experts often rely on up-to-date knowledge for apprehending and disseminating specific biological processes that help them design strategies to develop prevention and therapeutic decision-making. A challenging scenario for artificial intelligence (AI) is using biomedical data (e.g., texts, imaging, omics, and clinical) to provide diagnosis and treatment recommendations for cancerous conditions. Data and knowledge about cancer, drugs, genes, proteins, and their mechanism is spread across structured (knowledge bases (KBs)) and unstructured (e.g., scientific articles) sources. A large-scale knowledge graph (KG) can be constructed by integrating these data, followed by extracting facts about semantically interrelated entities and relations. Such KGs not only allow exploration and question answering (QA) but also allow domain experts to deduce new knowledge. However, exploring and querying large-scale KGs is tedious for non-domain users due to a lack of understanding of the underlying data assets and semantic technologies. In this paper, we develop a domain KG to leverage cancer-specific biomarker discovery and interactive QA. For this, a domain ontology called OncoNet Ontology (ONO) is developed to enable semantic reasoning for validating gene-disease relations. The KG is then enriched by harmonizing the ONO, controlled vocabularies, and additional biomedical concepts from scientific articles by employing BioBERT- and SciBERT-based information extraction (IE) methods. Further, since the biomedical domain is evolving, where new findings often replace old ones, without employing up-to-date findings, there is a high chance an AI system exhibits concept drift while providing diagnosis and treatment. Therefore, we finetuned the KG using large language models (LLMs) based on more recent articles and KBs that might not have been seen by the named entity recognition models.
PADME-SoSci: A Platform for Analytics and Distributed Machine Learning for the Social Sciences
Apr 03, 2023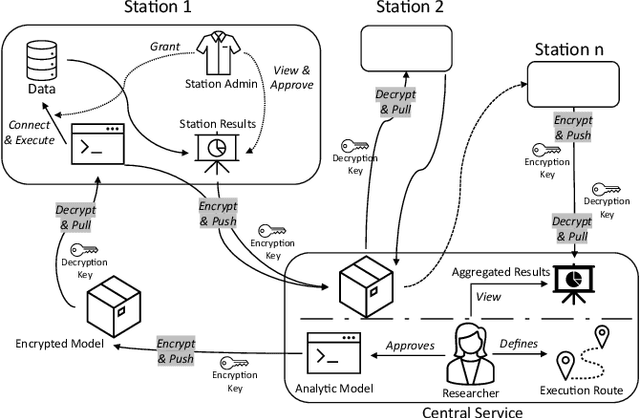
Data privacy and ownership are significant in social data science, raising legal and ethical concerns. Sharing and analyzing data is difficult when different parties own different parts of it. An approach to this challenge is to apply de-identification or anonymization techniques to the data before collecting it for analysis. However, this can reduce data utility and increase the risk of re-identification. To address these limitations, we present PADME, a distributed analytics tool that federates model implementation and training. PADME uses a federated approach where the model is implemented and deployed by all parties and visits each data location incrementally for training. This enables the analysis of data across locations while still allowing the model to be trained as if all data were in a single location. Training the model on data in its original location preserves data ownership. Furthermore, the results are not provided until the analysis is completed on all data locations to ensure privacy and avoid bias in the results.
Enhancing Data Space Semantic Interoperability through Machine Learning: a Visionary Perspective
Mar 15, 2023Our vision paper outlines a plan to improve the future of semantic interoperability in data spaces through the application of machine learning. The use of data spaces, where data is exchanged among members in a self-regulated environment, is becoming increasingly popular. However, the current manual practices of managing metadata and vocabularies in these spaces are time-consuming, prone to errors, and may not meet the needs of all stakeholders. By leveraging the power of machine learning, we believe that semantic interoperability in data spaces can be significantly improved. This involves automatically generating and updating metadata, which results in a more flexible vocabulary that can accommodate the diverse terminologies used by different sub-communities. Our vision for the future of data spaces addresses the limitations of conventional data exchange and makes data more accessible and valuable for all members of the community.
A Biomedical Knowledge Graph for Biomarker Discovery in Cancer
Feb 23, 2023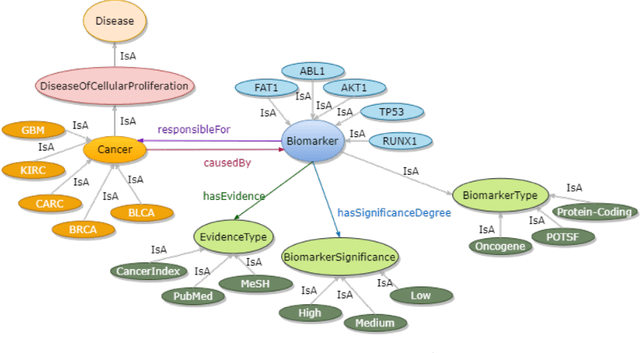
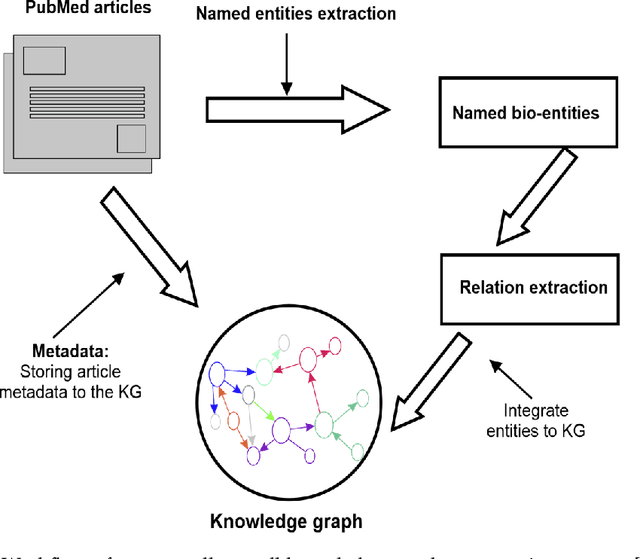

Structured and unstructured data and facts about drugs, genes, protein, viruses, and their mechanism are spread across a huge number of scientific articles. These articles are a large-scale knowledge source and can have a huge impact on disseminating knowledge about the mechanisms of certain biological processes. A domain-specific knowledge graph~(KG) is an explicit conceptualization of a specific subject-matter domain represented w.r.t semantically interrelated entities and relations. A KG can be constructed by integrating such facts and data and be used for data integration, exploration, and federated queries. However, exploration and querying large-scale KGs is tedious for certain groups of users due to a lack of knowledge about underlying data assets or semantic technologies. Such a KG will not only allow deducing new knowledge and question answering(QA) but also allows domain experts to explore. Since cross-disciplinary explanations are important for accurate diagnosis, it is important to query the KG to provide interactive explanations about learned biomarkers. Inspired by these, we construct a domain-specific KG, particularly for cancer-specific biomarker discovery. The KG is constructed by integrating cancer-related knowledge and facts from multiple sources. First, we construct a domain-specific ontology, which we call OncoNet Ontology (ONO). The ONO ontology is developed to enable semantic reasoning for verification of the predictions for relations between diseases and genes. The KG is then developed and enriched by harmonizing the ONO, additional metadata schemas, ontologies, controlled vocabularies, and additional concepts from external sources using a BERT-based information extraction method. BioBERT and SciBERT are finetuned with the selected articles crawled from PubMed. We listed down some queries and some examples of QA and deducing knowledge based on the KG.
Explainable AI for Bioinformatics: Methods, Tools, and Applications
Dec 25, 2022Artificial intelligence(AI) systems based on deep neural networks (DNNs) and machine learning (ML) algorithms are increasingly used to solve critical problems in bioinformatics, biomedical informatics, and precision medicine. However, complex DNN or ML models that are unavoidably opaque and perceived as black-box methods, may not be able to explain why and how they make certain decisions. Such black-box models are difficult to comprehend not only for targeted users and decision-makers but also for AI developers. Besides, in sensitive areas like healthcare, explainability and accountability are not only desirable properties of AI but also legal requirements -- especially when AI may have significant impacts on human lives. Explainable artificial intelligence (XAI) is an emerging field that aims to mitigate the opaqueness of black-box models and make it possible to interpret how AI systems make their decisions with transparency. An interpretable ML model can explain how it makes predictions and which factors affect the model's outcomes. The majority of state-of-the-art interpretable ML methods have been developed in a domain-agnostic way and originate from computer vision, automated reasoning, or even statistics. Many of these methods cannot be directly applied to bioinformatics problems, without prior customization, extension, and domain adoption. In this paper, we discuss the importance of explainability with a focus on bioinformatics. We analyse and comprehensively overview of model-specific and model-agnostic interpretable ML methods and tools. Via several case studies covering bioimaging, cancer genomics, and biomedical text mining, we show how bioinformatics research could benefit from XAI methods and how they could help improve decision fairness.
Towards General Deep Leakage in Federated Learning
Oct 18, 2021

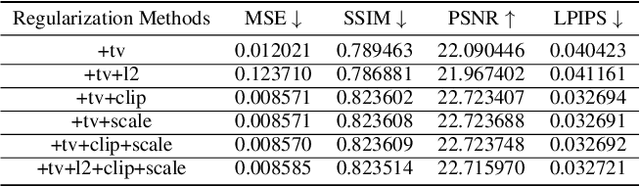
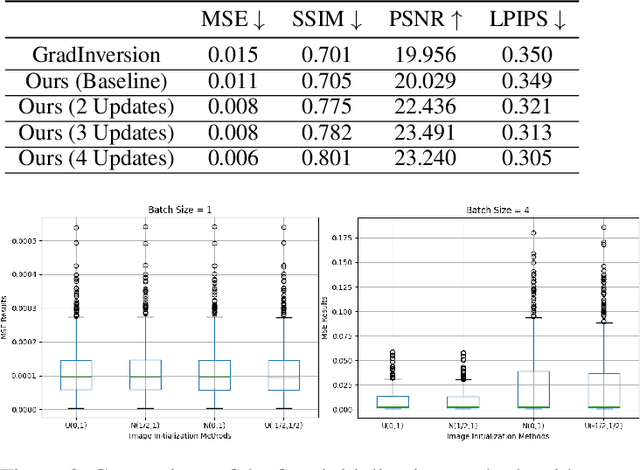
Unlike traditional central training, federated learning (FL) improves the performance of the global model by sharing and aggregating local models rather than local data to protect the users' privacy. Although this training approach appears secure, some research has demonstrated that an attacker can still recover private data based on the shared gradient information. This on-the-fly reconstruction attack deserves to be studied in depth because it can occur at any stage of training, whether at the beginning or at the end of model training; no relevant dataset is required and no additional models need to be trained. We break through some unrealistic assumptions and limitations to apply this reconstruction attack in a broader range of scenarios. We propose methods that can reconstruct the training data from shared gradients or weights, corresponding to the FedSGD and FedAvg usage scenarios, respectively. We propose a zero-shot approach to restore labels even if there are duplicate labels in the batch. We study the relationship between the label and image restoration. We find that image restoration fails even if there is only one incorrectly inferred label in the batch; we also find that when batch images have the same label, the corresponding image is restored as a fusion of that class of images. Our approaches are evaluated on classic image benchmarks, including CIFAR-10 and ImageNet. The batch size, image quality, and the adaptability of the label distribution of our approach exceed those of GradInversion, the state-of-the-art.
DeepCOVIDExplainer: Explainable COVID-19 Predictions Based on Chest X-ray Images
Apr 10, 2020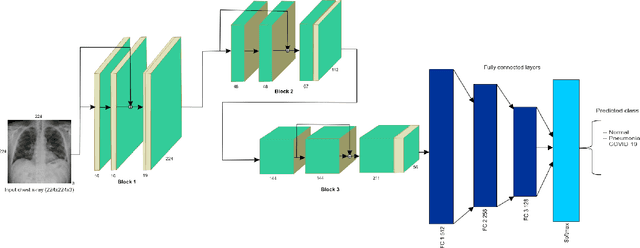
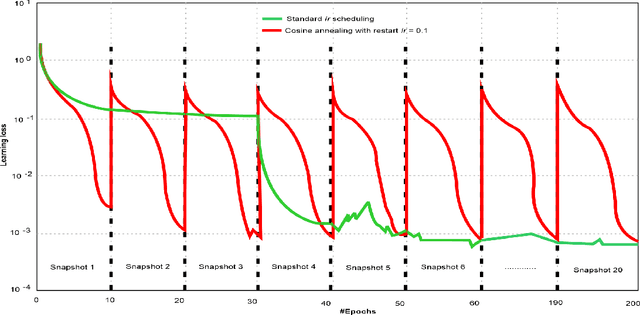
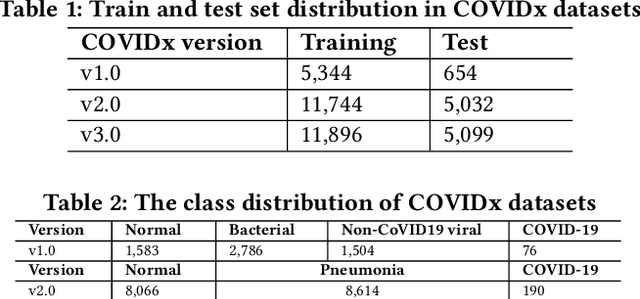
Amid the coronavirus disease(COVID-19) pandemic, humanity experiences a rapid increase in infection numbers across the world. Challenge hospitals are faced with, in the fight against the virus, is the effective screening of incoming patients. One methodology is the assessment of chest radiography(CXR) images, which usually requires expert radiologists' knowledge. In this paper, we propose an explainable deep neural networks(DNN)-based method for automatic detection of COVID-19 symptoms from CXR images, which we call 'DeepCOVIDExplainer'. We used 16,995 CXR images across 13,808 patients, covering normal, pneumonia, and COVID-19 cases. CXR images are first comprehensively preprocessed, before being augmented and classified with a neural ensemble method, followed by highlighting class-discriminating regions using gradient-guided class activation maps(Grad-CAM++) and layer-wise relevance propagation(LRP). Further, we provide human-interpretable explanations of the predictions. Evaluation results based on hold-out data show that our approach can identify COVID-19 confidently with a positive predictive value(PPV) of 89.61% and recall of 83%, improving over recent comparable approaches. We hope that our findings will be a useful contribution to the fight against COVID-19 and, in more general, towards an increasing acceptance and adoption of AI-assisted applications in the clinical practice.
OncoNetExplainer: Explainable Predictions of Cancer Types Based on Gene Expression Data
Sep 09, 2019
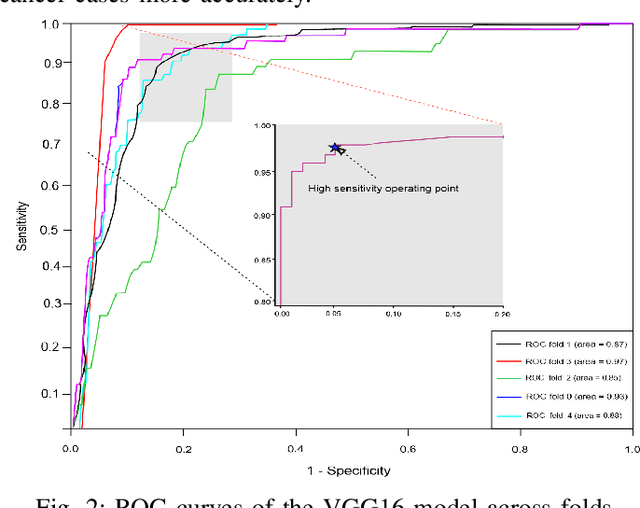

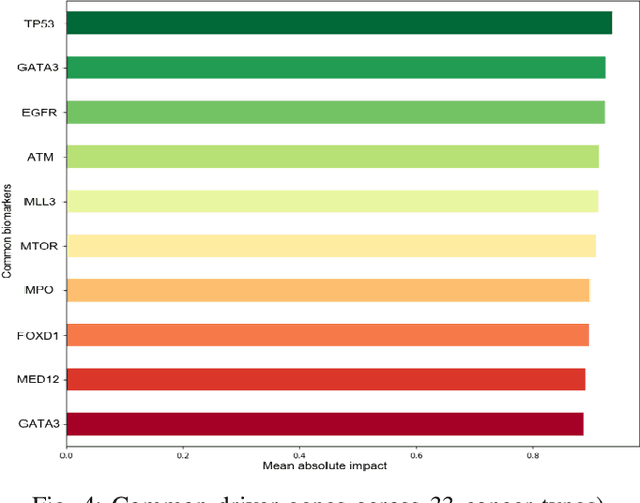
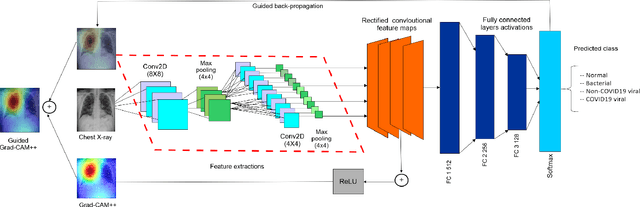
The discovery of important biomarkers is a significant step towards understanding the molecular mechanisms of carcinogenesis; enabling accurate diagnosis for, and prognosis of, a certain cancer type. Before recommending any diagnosis, genomics data such as gene expressions(GE) and clinical outcomes need to be analyzed. However, complex nature, high dimensionality, and heterogeneity in genomics data make the overall analysis challenging. Convolutional neural networks(CNN) have shown tremendous success in solving such problems. However, neural network models are perceived mostly as `black box' methods because of their not well-understood internal functioning. However, interpretability is important to provide insights on why a given cancer case has a certain type. Besides, finding the most important biomarkers can help in recommending more accurate treatments and drug repositioning. In this paper, we propose a new approach called OncoNetExplainer to make explainable predictions of cancer types based on GE data. We used genomics data about 9,074 cancer patients covering 33 different cancer types from the Pan-Cancer Atlas on which we trained CNN and VGG16 networks using guided-gradient class activation maps++(GradCAM++). Further, we generate class-specific heat maps to identify significant biomarkers and computed feature importance in terms of mean absolute impact to rank top genes across all the cancer types. Quantitative and qualitative analyses show that both models exhibit high confidence at predicting the cancer types correctly giving an average precision of 96.25%. To provide comparisons with the baselines, we identified top genes, and cancer-specific driver genes using gradient boosted trees and SHapley Additive exPlanations(SHAP). Finally, our findings were validated with the annotations provided by the TumorPortal.
* In proc. of 19th IEEE International Conference on Bioinformatics and Bioengineering(IEEE BIBE 2019)
Drug-Drug Interaction Prediction Based on Knowledge Graph Embeddings and Convolutional-LSTM Network
Aug 04, 2019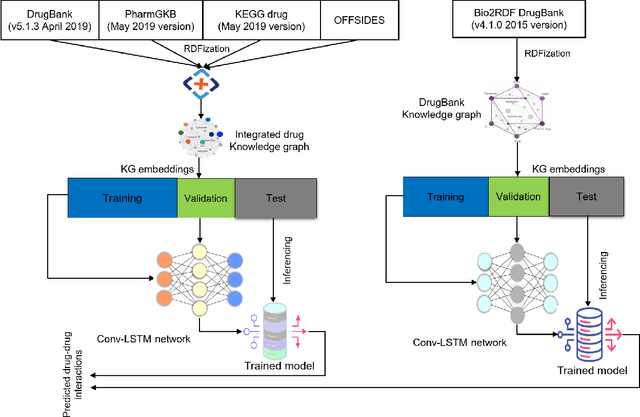

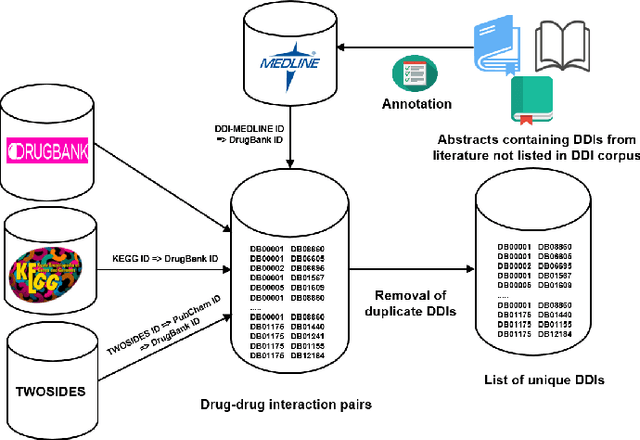
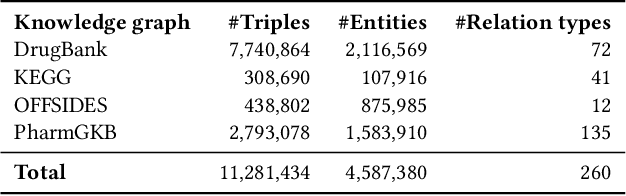
Interference between pharmacological substances can cause serious medical injuries. Correctly predicting so-called drug-drug interactions (DDI) does not only reduce these cases but can also result in a reduction of drug development cost. Presently, most drug-related knowledge is the result of clinical evaluations and post-marketing surveillance; resulting in a limited amount of information. Existing data-driven prediction approaches for DDIs typically rely on a single source of information, while using information from multiple sources would help improve predictions. Machine learning (ML) techniques are used, but the techniques are often unable to deal with skewness in the data. Hence, we propose a new ML approach for predicting DDIs based on multiple data sources. For this task, we use 12,000 drug features from DrugBank, PharmGKB, and KEGG drugs, which are integrated using Knowledge Graphs (KGs). To train our prediction model, we first embed the nodes in the graph using various embedding approaches. We found that the best performing combination was a ComplEx embedding method creating using PyTorch-BigGraph (PBG) with a Convolutional-LSTM network and classic machine learning-based prediction models. The model averaging ensemble method of three best classifiers yields up to 0.94, 0.92, 0.80 for AUPR, F1-score, and MCC, respectively during 5-fold cross-validation tests.