 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFinetuning-Free Diffusion Model with Adaptive Constraint Guidance for Inorganic Crystal Structure Generation
Apr 14, 2026The discovery of inorganic crystal structures with targeted properties is a significant challenge in materials science. Generative models, especially state-of-the-art diffusion models, offer the promise of modeling complex data distributions and proposing novel, realistic samples. However, current generative AI models still struggle to produce diverse, original, and reliable structures of experimentally achievable materials suitable for high-stakes applications. In this work, we propose a generative machine learning framework based on diffusion models with adaptive constraint guidance, which enables the incorporation of user-defined physical and chemical constraints during the generation process. This approach is designed to be practical and interpretable for human experts, allowing transparent decision-making and expert-driven exploration. To ensure the robustness and validity of the generated candidates, we introduce a multi-step validation pipeline that combines graph neural network estimators trained to achieve DFT-level accuracy and convex hull analysis for assessing thermodynamic stability. Our approach has been tested and validated on several classical examples of inorganic families of compounds, as case studies. As a consequence, these preliminary results demonstrate our framework's ability to generate thermodynamically plausible crystal structures that satisfy targeted geometric constraints across diverse inorganic chemical systems.
Bottom-up Iterative Anomalous Diffusion Detector (BI-ADD)
Mar 14, 2025In recent years, the segmentation of short molecular trajectories with varying diffusive properties has drawn particular attention of researchers, since it allows studying the dynamics of a particle. In the past decade, machine learning methods have shown highly promising results, also in changepoint detection and segmentation tasks. Here, we introduce a novel iterative method to identify the changepoints in a molecular trajectory, i.e., frames, where the diffusive behavior of a particle changes. A trajectory in our case follows a fractional Brownian motion and we estimate the diffusive properties of the trajectories. The proposed BI-ADD combines unsupervised and supervised learning methods to detect the changepoints. Our approach can be used for the analysis of molecular trajectories at the individual level and also be extended to multiple particle tracking, which is an important challenge in fundamental biology. We validated BI-ADD in various scenarios within the framework of the AnDi2 Challenge 2024 dedicated to single particle tracking. Our method is implemented in Python and is publicly available for research purposes.
Data-Driven Score-Based Models for Generating Stable Structures with Adaptive Crystal Cells
Oct 16, 2023The discovery of new functional and stable materials is a big challenge due to its complexity. This work aims at the generation of new crystal structures with desired properties, such as chemical stability and specified chemical composition, by using machine learning generative models. Compared to the generation of molecules, crystal structures pose new difficulties arising from the periodic nature of the crystal and from the specific symmetry constraints related to the space group. In this work, score-based probabilistic models based on annealed Langevin dynamics, which have shown excellent performance in various applications, are adapted to the task of crystal generation. The novelty of the presented approach resides in the fact that the lattice of the crystal cell is not fixed. During the training of the model, the lattice is learned from the available data, whereas during the sampling of a new chemical structure, two denoising processes are used in parallel to generate the lattice along the generation of the atomic positions. A multigraph crystal representation is introduced that respects symmetry constraints, yielding computational advantages and a better quality of the sampled structures. We show that our model is capable of generating new candidate structures in any chosen chemical system and crystal group without any additional training. To illustrate the functionality of the proposed method, a comparison of our model to other recent generative models, based on descriptor-based metrics, is provided.
False clustering rate control in mixture models
Mar 08, 2022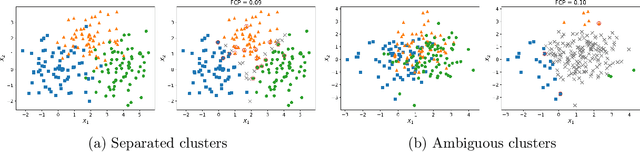
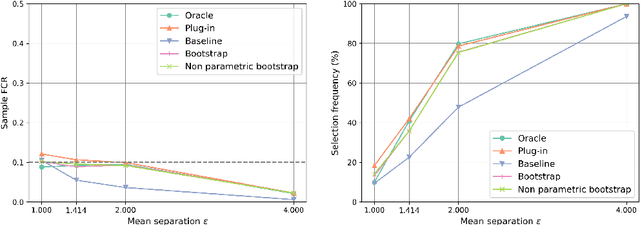
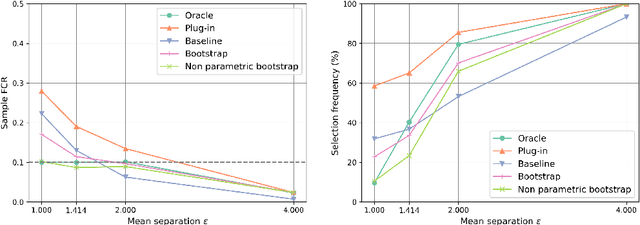
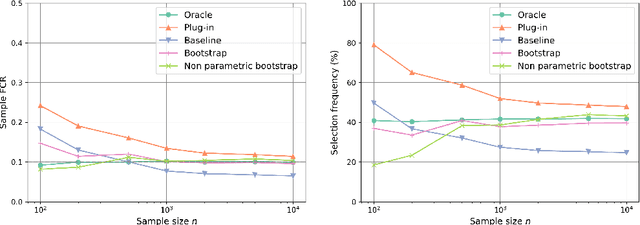
The clustering task consists in delivering labels to the members of a sample. For most data sets, some individuals are ambiguous and intrinsically difficult to attribute to one or another cluster. However, in practical applications, misclassifying individuals is potentially disastrous. To overcome this difficulty, the idea followed here is to classify only a part of the sample in order to obtain a small misclassification rate. This approach is well known in the supervised setting, and referred to as classification with an abstention option. The purpose of this paper is to revisit this approach in an unsupervised mixture-model framework. The problem is formalized in terms of controlling the false clustering rate (FCR) below a prescribed level {\alpha}, while maximizing the number of classified items. New procedures are introduced and their behavior is shown to be close to the optimal one by establishing theoretical results and conducting numerical experiments. An application to breast cancer data illustrates the benefits of the new approach from a practical viewpoint.
Supervised deep learning prediction of the formation enthalpy of the full set of configurations in complex phases: the $σ-$phase as an example
Nov 21, 2020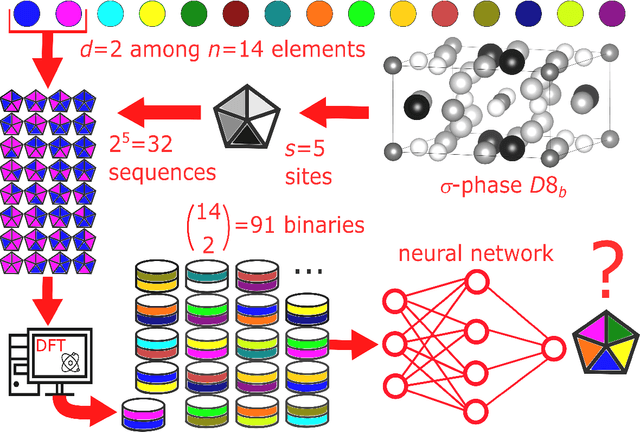
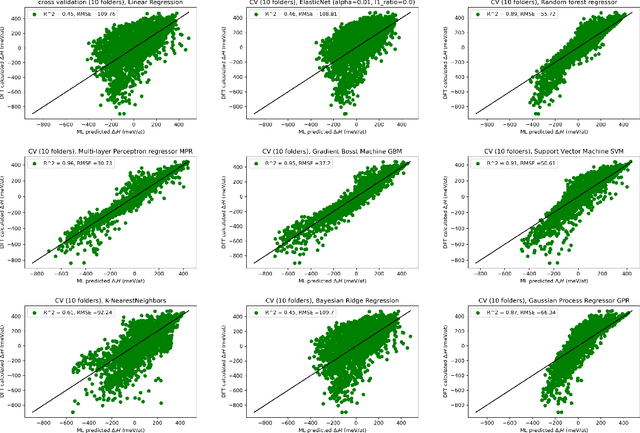
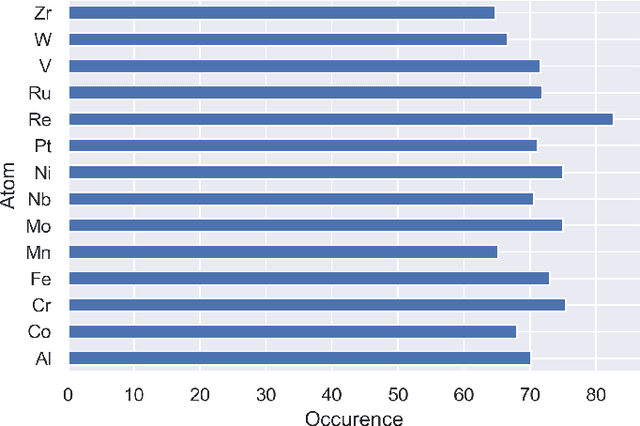
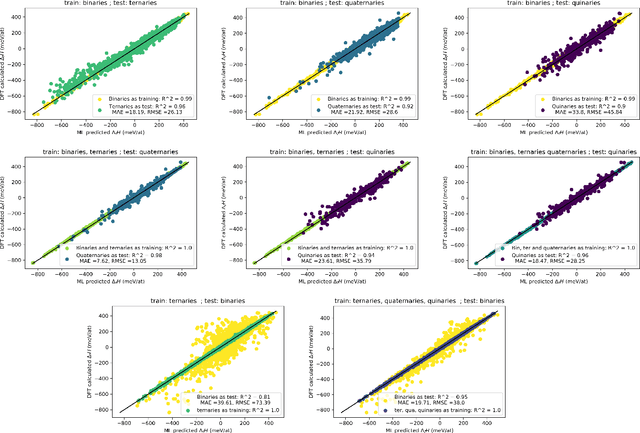
Machine learning (ML) methods are becoming integral to scientific inquiry in numerous disciplines, such as material sciences. In this manuscript, we demonstrate how ML can be used to predict several properties in solid-state chemistry, in particular the heat of formation of a given complex crystallographic phase (here the $\sigma-$phase, $tP30$, $D8_{b}$). Based on an independent and unprecedented large first principles dataset containing about 10,000 $\sigma-$compounds with $n=14$ different elements, we used a supervised learning approach, to predict all the $\sim$500,000 possible configurations within a mean absolute error of 23 meV/at ($\sim$2 kJ.mol$^{-1}$) on the heat of formation and $\sim$0.06 Ang. on the tetragonal cell parameters. We showed that neural network regression algorithms provide a significant improvement in accuracy of the predicted output compared to traditional regression techniques. Adding descriptors having physical nature (atomic radius, number of valence electrons) improves the learning precision. Based on our analysis, the training database composed of the only binary-compositions plays a major role in predicting the higher degree system configurations. Our result opens a broad avenue to efficient high-throughput investigations of the combinatorial binary calculation for multicomponent prediction of a complex phase.
Latent Instrumental Variables as Priors in Causal Inference based on Independence of Cause and Mechanism
Jul 17, 2020
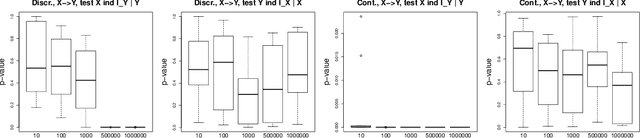

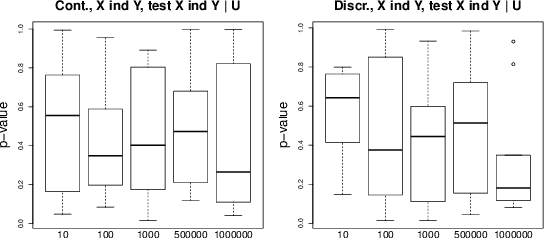
Causal inference methods based on conditional independence construct Markov equivalent graphs, and cannot be applied to bivariate cases. The approaches based on independence of cause and mechanism state, on the contrary, that causal discovery can be inferred for two observations. In our contribution, we challenge to reconcile these two research directions. We study the role of latent variables such as latent instrumental variables and hidden common causes in the causal graphical structures. We show that the methods based on the independence of cause and mechanism, indirectly contain traces of the existence of the hidden instrumental variables. We derive a novel algorithm to infer causal relationships between two variables, and we validate the proposed method on simulated data and on a benchmark of cause-effect pairs. We illustrate by our experiments that the proposed approach is simple and extremely competitive in terms of empirical accuracy compared to the state-of-the-art methods.
CrystalGAN: Learning to Discover Crystallographic Structures with Generative Adversarial Networks
Oct 26, 2018
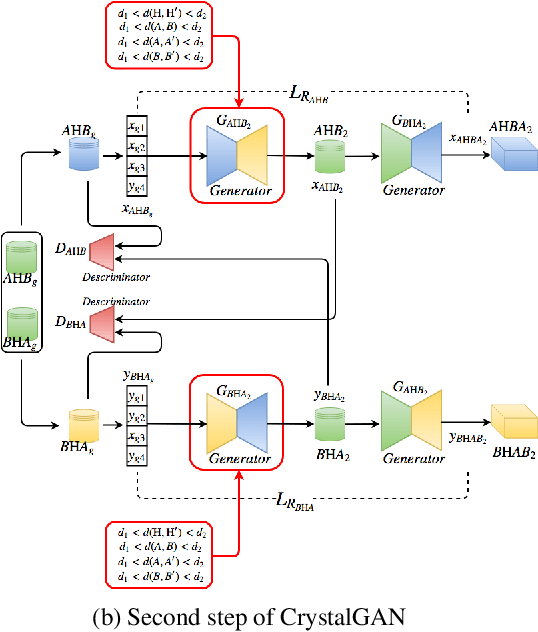
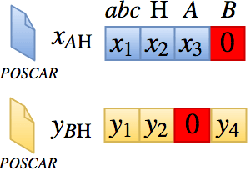
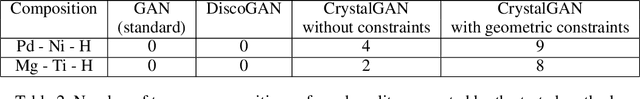
Our main motivation is to propose an efficient approach to generate novel multi-element stable chemical compounds that can be used in real world applications. This task can be formulated as a combinatorial problem, and it takes many hours of human experts to construct, and to evaluate new data. Unsupervised learning methods such as Generative Adversarial Networks (GANs) can be efficiently used to produce new data. Cross-domain Generative Adversarial Networks were reported to achieve exciting results in image processing applications. However, in the domain of materials science, there is a need to synthesize data with higher order complexity compared to observed samples, and the state-of-the-art cross-domain GANs can not be adapted directly. In this contribution, we propose a novel GAN called CrystalGAN which generates new chemically stable crystallographic structures with increased domain complexity. We introduce an original architecture, we provide the corresponding loss functions, and we show that the CrystalGAN generates very reasonable data. We illustrate the efficiency of the proposed method on a real original problem of novel hydrides discovery that can be further used in development of hydrogen storage materials.
Disease Classification in Metagenomics with 2D Embeddings and Deep Learning
Jun 23, 2018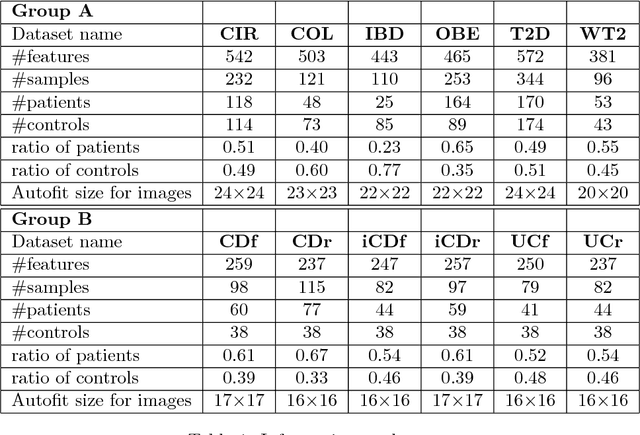
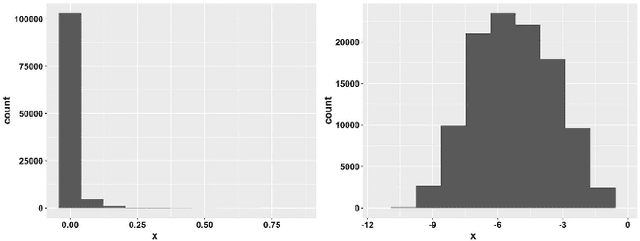
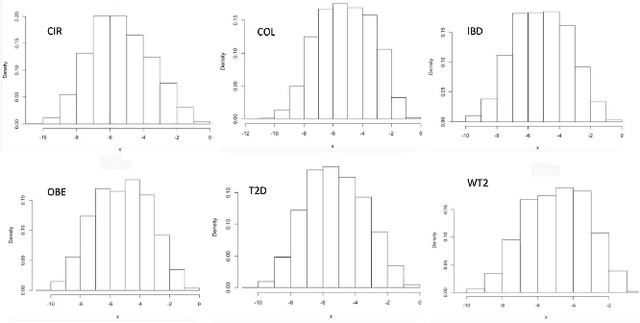
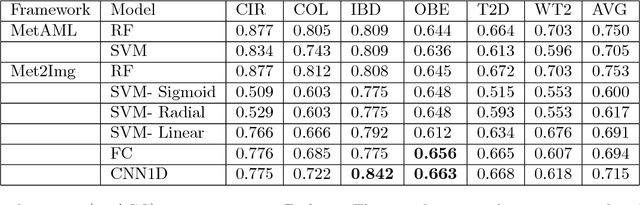
Deep learning (DL) techniques have shown unprecedented success when applied to images, waveforms, and text. Generally, when the sample size ($N$) is much bigger than the number of features ($d$), DL often outperforms other machine learning (ML) techniques, often through the use of Convolutional Neural Networks (CNNs). However, in many bioinformatics fields (including metagenomics), we encounter the opposite situation where $d$ is significantly greater than $N$. In these situations, applying DL techniques would lead to severe overfitting. Here we aim to improve classification of various diseases with metagenomic data through the use of CNNs. For this we proposed to represent metagenomic data as images. The proposed Met2Img approach relies on taxonomic and t-SNE embeddings to transform abundance data into "synthetic images". We applied our approach to twelve benchmark data sets including more than 1400 metagenomic samples. Our results show significant improvements over the state-of-the-art algorithms (Random Forest (RF), Support Vector Machine (SVM)). We observe that the integration of phylogenetic information alongside abundance data improves classification. The proposed approach is not only important in classification setting but also allows to visualize complex metagenomic data. The Met2Img is implemented in Python.
Deep Learning for Metagenomic Data: using 2D Embeddings and Convolutional Neural Networks
Dec 01, 2017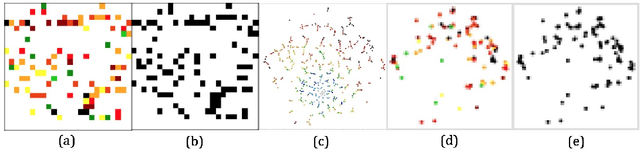
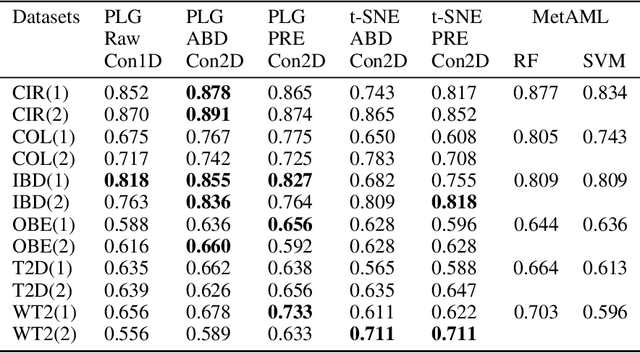
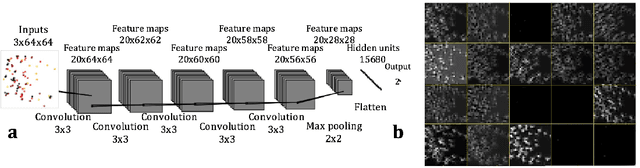
Deep learning (DL) techniques have had unprecedented success when applied to images, waveforms, and texts to cite a few. In general, when the sample size (N) is much greater than the number of features (d), DL outperforms previous machine learning (ML) techniques, often through the use of convolution neural networks (CNNs). However, in many bioinformatics ML tasks, we encounter the opposite situation where d is greater than N. In these situations, applying DL techniques (such as feed-forward networks) would lead to severe overfitting. Thus, sparse ML techniques (such as LASSO e.g.) usually yield the best results on these tasks. In this paper, we show how to apply CNNs on data which do not have originally an image structure (in particular on metagenomic data). Our first contribution is to show how to map metagenomic data in a meaningful way to 1D or 2D images. Based on this representation, we then apply a CNN, with the aim of predicting various diseases. The proposed approach is applied on six different datasets including in total over 1000 samples from various diseases. This approach could be a promising one for prediction tasks in the bioinformatics field.
Efficient Learning of Sparse Conditional Random Fields for Supervised Sequence Labelling
Jan 03, 2010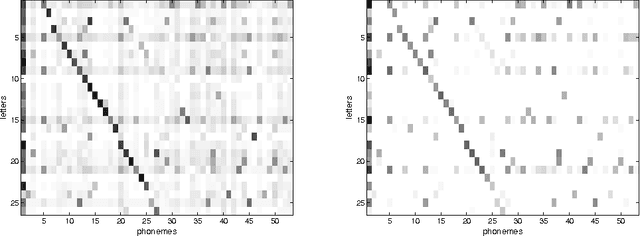

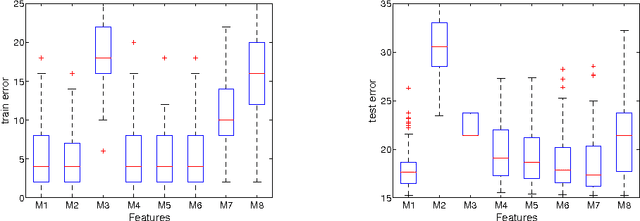
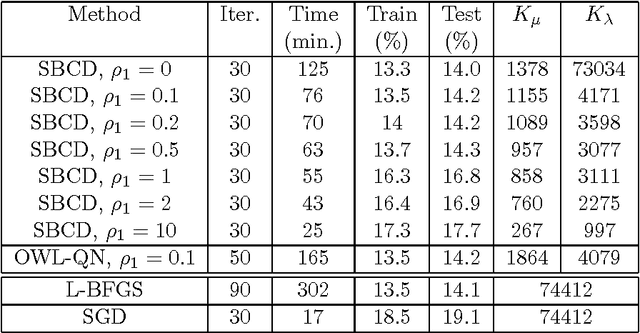
Conditional Random Fields (CRFs) constitute a popular and efficient approach for supervised sequence labelling. CRFs can cope with large description spaces and can integrate some form of structural dependency between labels. In this contribution, we address the issue of efficient feature selection for CRFs based on imposing sparsity through an L1 penalty. We first show how sparsity of the parameter set can be exploited to significantly speed up training and labelling. We then introduce coordinate descent parameter update schemes for CRFs with L1 regularization. We finally provide some empirical comparisons of the proposed approach with state-of-the-art CRF training strategies. In particular, it is shown that the proposed approach is able to take profit of the sparsity to speed up processing and hence potentially handle larger dimensional models.