 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMetagenBERT: a Transformer-based Architecture using Foundational genomic Large Language Models for novel Metagenome Representation
Jan 05, 2026Metagenomic disease prediction commonly relies on species abundance tables derived from large, incomplete reference catalogs, constraining resolution and discarding valuable information contained in DNA reads. To overcome these limitations, we introduce MetagenBERT, a Transformer based framework that produces end to end metagenome embeddings directly from raw DNA sequences, without taxonomic or functional annotations. Reads are embedded using foundational genomic language models (DNABERT2 and the microbiome specialized DNABERTMS), then aggregated through a scalable clustering strategy based on FAISS accelerated KMeans. Each metagenome is represented as a cluster abundance vector summarizing the distribution of its embedded reads. We evaluate this approach on five benchmark gut microbiome datasets (Cirrhosis, T2D, Obesity, IBD, CRC). MetagenBERT achieves competitive or superior AUC performance relative to species abundance baselines across most tasks. Concatenating both representations further improves prediction, demonstrating complementarity between taxonomic and embedding derived signals. Clustering remains robust when applied to as little as 10% of reads, highlighting substantial redundancy in metagenomes and enabling major computational gains. We additionally introduce MetagenBERT Glob Mcardis, a cross cohort variant trained on the large, phenotypically diverse MetaCardis cohort and transferred to other datasets, retaining predictive signal including for unseen phenotypes, indicating the feasibility of a foundation model for metagenome representation. Robustness analyses (PERMANOVA, PERMDISP, entropy) show consistent separation of different states across subsamples. Overall, MetagenBERT provides a scalable, annotation free representation of metagenomes pointing toward future phenotype aware generalization across heterogeneous cohorts and sequencing technologies.
PlantSAM: An Object Detection-Driven Segmentation Pipeline for Herbarium Specimens
Jul 22, 2025Deep learning-based classification of herbarium images is hampered by background heterogeneity, which introduces noise and artifacts that can potentially mislead models and reduce classification accuracy. Addressing these background-related challenges is critical to improving model performance. We introduce PlantSAM, an automated segmentation pipeline that integrates YOLOv10 for plant region detection and the Segment Anything Model (SAM2) for segmentation. YOLOv10 generates bounding box prompts to guide SAM2, enhancing segmentation accuracy. Both models were fine-tuned on herbarium images and evaluated using Intersection over Union (IoU) and Dice coefficient metrics. PlantSAM achieved state-of-the-art segmentation performance, with an IoU of 0.94 and a Dice coefficient of 0.97. Incorporating segmented images into classification models led to consistent performance improvements across five tested botanical traits, with accuracy gains of up to 4.36% and F1-score improvements of 4.15%. Our findings highlight the importance of background removal in herbarium image analysis, as it significantly enhances classification accuracy by allowing models to focus more effectively on the foreground plant structures.
IKrNet: A Neural Network for Detecting Specific Drug-Induced Patterns in Electrocardiograms Amidst Physiological Variability
May 12, 2025Monitoring and analyzing electrocardiogram (ECG) signals, even under varying physiological conditions, including those influenced by physical activity, drugs and stress, is crucial to accurately assess cardiac health. However, current AI-based methods often fail to account for how these factors interact and alter ECG patterns, ultimately limiting their applicability in real-world settings. This study introduces IKrNet, a novel neural network model, which identifies drug-specific patterns in ECGs amidst certain physiological conditions. IKrNet's architecture incorporates spatial and temporal dynamics by using a convolutional backbone with varying receptive field size to capture spatial features. A bi-directional Long Short-Term Memory module is also employed to model temporal dependencies. By treating heart rate variability as a surrogate for physiological fluctuations, we evaluated IKrNet's performance across diverse scenarios, including conditions with physical stress, drug intake alone, and a baseline without drug presence. Our assessment follows a clinical protocol in which 990 healthy volunteers were administered 80mg of Sotalol, a drug which is known to be a precursor to Torsades-de-Pointes, a life-threatening arrhythmia. We show that IKrNet outperforms state-of-the-art models' accuracy and stability in varying physiological conditions, underscoring its clinical viability.
Disease Classification in Metagenomics with 2D Embeddings and Deep Learning
Jun 23, 2018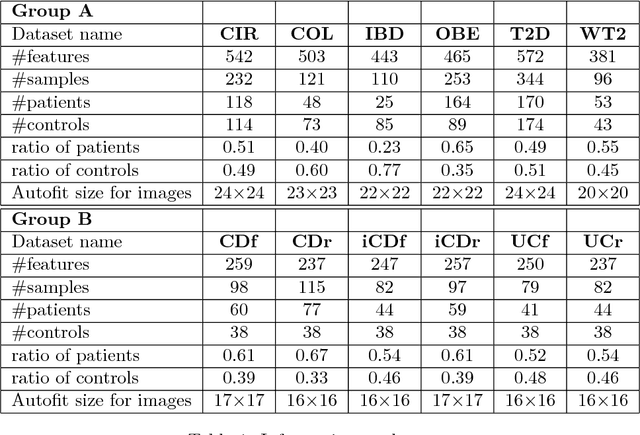
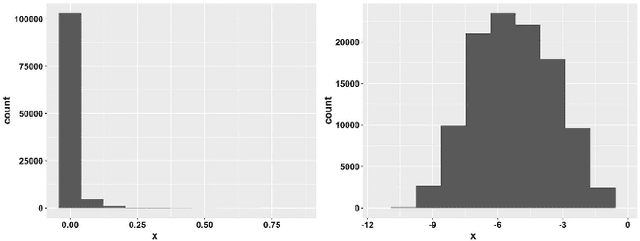
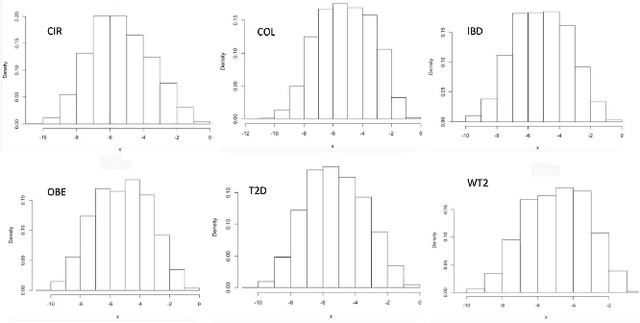
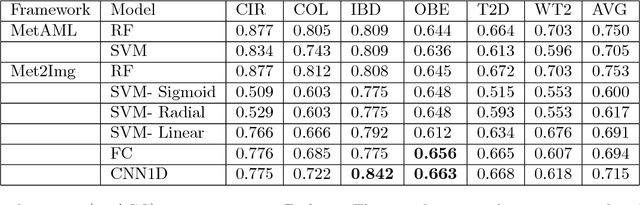
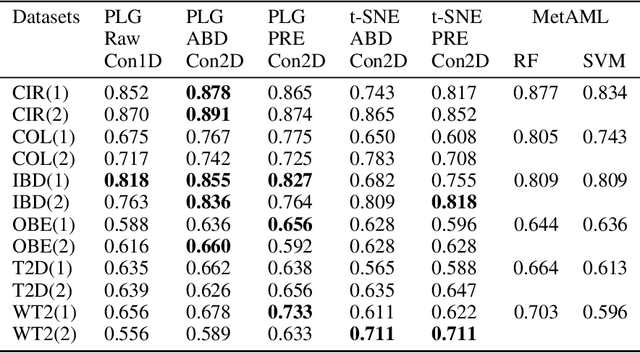
Deep learning (DL) techniques have shown unprecedented success when applied to images, waveforms, and text. Generally, when the sample size ($N$) is much bigger than the number of features ($d$), DL often outperforms other machine learning (ML) techniques, often through the use of Convolutional Neural Networks (CNNs). However, in many bioinformatics fields (including metagenomics), we encounter the opposite situation where $d$ is significantly greater than $N$. In these situations, applying DL techniques would lead to severe overfitting. Here we aim to improve classification of various diseases with metagenomic data through the use of CNNs. For this we proposed to represent metagenomic data as images. The proposed Met2Img approach relies on taxonomic and t-SNE embeddings to transform abundance data into "synthetic images". We applied our approach to twelve benchmark data sets including more than 1400 metagenomic samples. Our results show significant improvements over the state-of-the-art algorithms (Random Forest (RF), Support Vector Machine (SVM)). We observe that the integration of phylogenetic information alongside abundance data improves classification. The proposed approach is not only important in classification setting but also allows to visualize complex metagenomic data. The Met2Img is implemented in Python.
Deep Learning for Metagenomic Data: using 2D Embeddings and Convolutional Neural Networks
Dec 01, 2017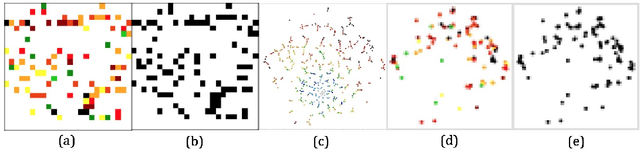
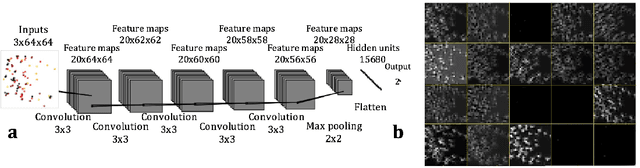
Deep learning (DL) techniques have had unprecedented success when applied to images, waveforms, and texts to cite a few. In general, when the sample size (N) is much greater than the number of features (d), DL outperforms previous machine learning (ML) techniques, often through the use of convolution neural networks (CNNs). However, in many bioinformatics ML tasks, we encounter the opposite situation where d is greater than N. In these situations, applying DL techniques (such as feed-forward networks) would lead to severe overfitting. Thus, sparse ML techniques (such as LASSO e.g.) usually yield the best results on these tasks. In this paper, we show how to apply CNNs on data which do not have originally an image structure (in particular on metagenomic data). Our first contribution is to show how to map metagenomic data in a meaningful way to 1D or 2D images. Based on this representation, we then apply a CNN, with the aim of predicting various diseases. The proposed approach is applied on six different datasets including in total over 1000 samples from various diseases. This approach could be a promising one for prediction tasks in the bioinformatics field.