 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSpecifying and Reasoning about CPS through the Lens of the NIST CPS Framework
Jan 14, 2022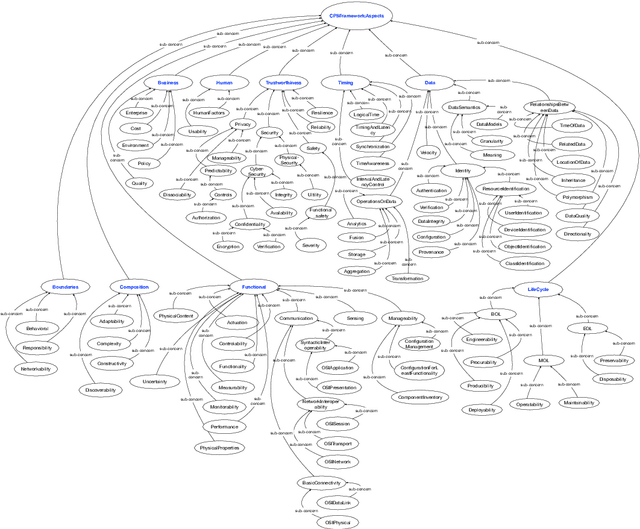
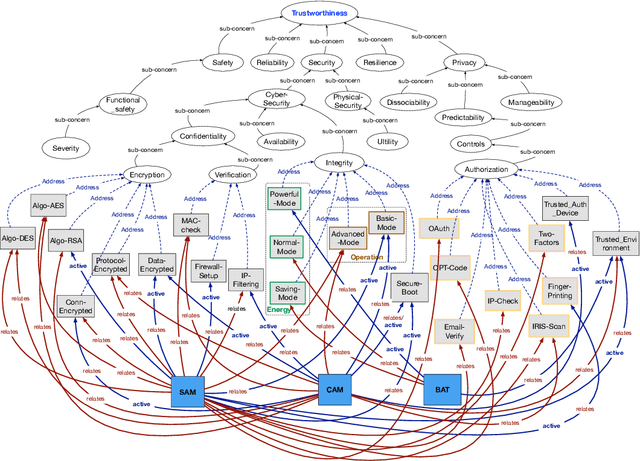

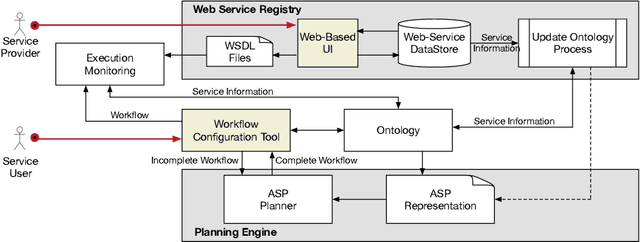
This paper introduces a formal definition of a Cyber-Physical System (CPS) in the spirit of the CPS Framework proposed by the National Institute of Standards and Technology (NIST). It shows that using this definition, various problems related to concerns in a CPS can be precisely formalized and implemented using Answer Set Programming (ASP). These include problems related to the dependency or conflicts between concerns, how to mitigate an issue, and what the most suitable mitigation strategy for a given issue would be. It then shows how ASP can be used to develop an implementation that addresses the aforementioned problems. The paper concludes with a discussion of the potentials of the proposed methodologies.
Disease Classification in Metagenomics with 2D Embeddings and Deep Learning
Jun 23, 2018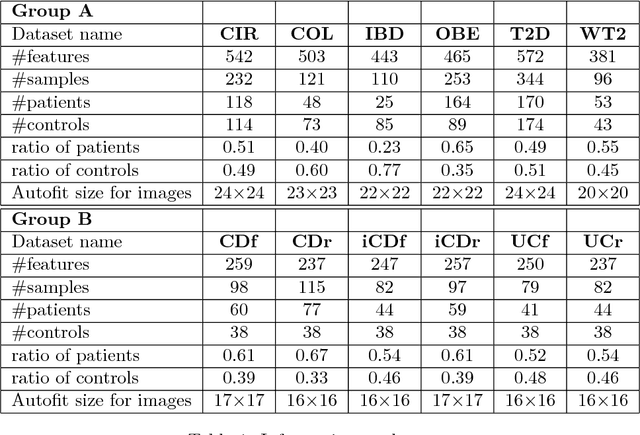
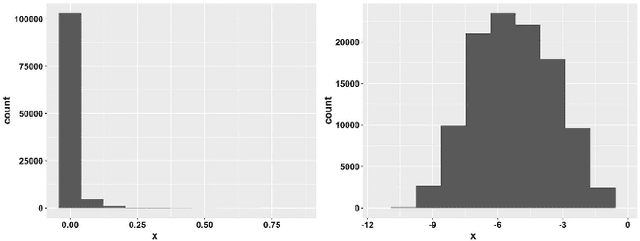
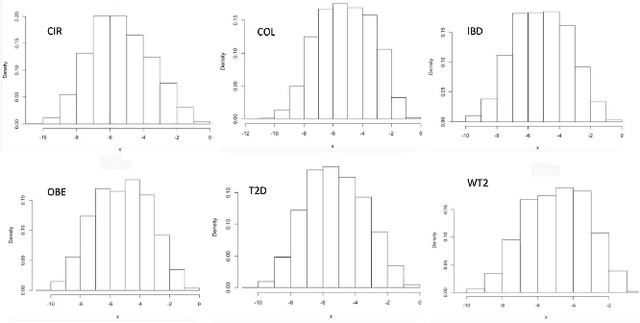
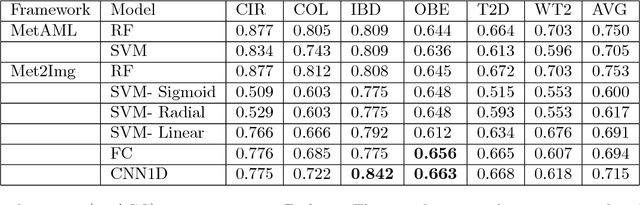
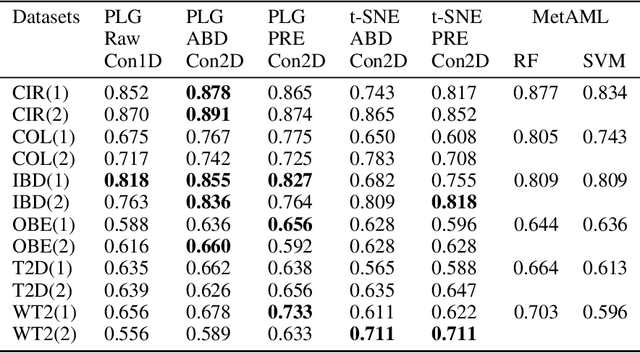
Deep learning (DL) techniques have shown unprecedented success when applied to images, waveforms, and text. Generally, when the sample size ($N$) is much bigger than the number of features ($d$), DL often outperforms other machine learning (ML) techniques, often through the use of Convolutional Neural Networks (CNNs). However, in many bioinformatics fields (including metagenomics), we encounter the opposite situation where $d$ is significantly greater than $N$. In these situations, applying DL techniques would lead to severe overfitting. Here we aim to improve classification of various diseases with metagenomic data through the use of CNNs. For this we proposed to represent metagenomic data as images. The proposed Met2Img approach relies on taxonomic and t-SNE embeddings to transform abundance data into "synthetic images". We applied our approach to twelve benchmark data sets including more than 1400 metagenomic samples. Our results show significant improvements over the state-of-the-art algorithms (Random Forest (RF), Support Vector Machine (SVM)). We observe that the integration of phylogenetic information alongside abundance data improves classification. The proposed approach is not only important in classification setting but also allows to visualize complex metagenomic data. The Met2Img is implemented in Python.
Phylotastic: An Experiment in Creating, Manipulating, and Evolving Phylogenetic Biology Workflows Using Logic Programming
May 01, 2018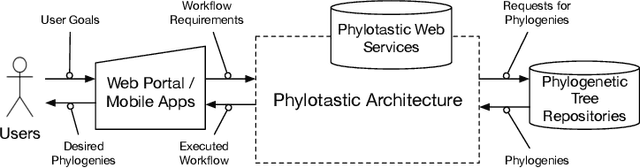
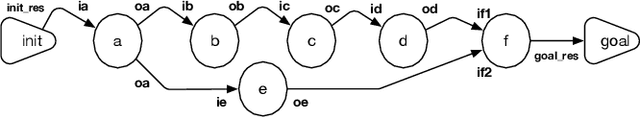
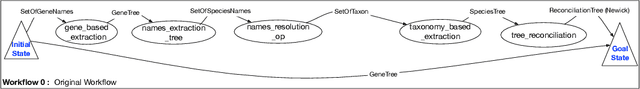
Evolutionary Biologists have long struggled with the challenge of developing analysis workflows in a flexible manner, thus facilitating the reuse of phylogenetic knowledge. An evolutionary biology workflow can be viewed as a plan which composes web services that can retrieve, manipulate, and produce phylogenetic trees. The Phylotastic project was launched two years ago as a collaboration between evolutionary biologists and computer scientists, with the goal of developing an open architecture to facilitate the creation of such analysis workflows. While composition of web services is a problem that has been extensively explored in the literature, including within the logic programming domain, the incarnation of the problem in Phylotastic provides a number of additional challenges. Along with the need to integrate preferences and formal ontologies in the description of the desired workflow, evolutionary biologists tend to construct workflows in an incremental manner, by successively refining the workflow, by indicating desired changes (e.g., exclusion of certain services, modifications of the desired output). This leads to the need of successive iterations of incremental replanning, to develop a new workflow that integrates the requested changes while minimizing the changes to the original workflow. This paper illustrates how Phylotastic has addressed the challenges of creating and refining phylogenetic analysis workflows using logic programming technology and how such solutions have been used within the general framework of the Phylotastic project. Under consideration in Theory and Practice of Logic Programming (TPLP).
Deep Learning for Metagenomic Data: using 2D Embeddings and Convolutional Neural Networks
Dec 01, 2017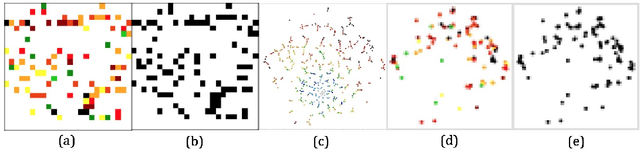
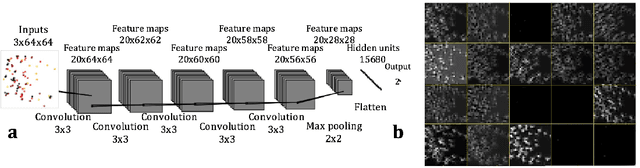
Deep learning (DL) techniques have had unprecedented success when applied to images, waveforms, and texts to cite a few. In general, when the sample size (N) is much greater than the number of features (d), DL outperforms previous machine learning (ML) techniques, often through the use of convolution neural networks (CNNs). However, in many bioinformatics ML tasks, we encounter the opposite situation where d is greater than N. In these situations, applying DL techniques (such as feed-forward networks) would lead to severe overfitting. Thus, sparse ML techniques (such as LASSO e.g.) usually yield the best results on these tasks. In this paper, we show how to apply CNNs on data which do not have originally an image structure (in particular on metagenomic data). Our first contribution is to show how to map metagenomic data in a meaningful way to 1D or 2D images. Based on this representation, we then apply a CNN, with the aim of predicting various diseases. The proposed approach is applied on six different datasets including in total over 1000 samples from various diseases. This approach could be a promising one for prediction tasks in the bioinformatics field.