 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMaximum Entropy Relaxation of Multi-Way Cardinality Constraints for Synthetic Population Generation
Mar 23, 2026Generating synthetic populations from aggregate statistics is a core component of microsimulation, agent-based modeling, policy analysis, and privacy-preserving data release. Beyond classical census marginals, many applications require matching heterogeneous unary, binary, and ternary constraints derived from surveys, expert knowledge, or automatically extracted descriptions. Constructing populations that satisfy such multi-way constraints simultaneously poses a significant computational challenge. We consider populations where each individual is described by categorical attributes and the target is a collection of global frequency constraints over attribute combinations. Exact formulations scale poorly as the number and arity of constraints increase, especially when the constraints are numerous and overlapping. Grounded in methods from statistical physics, we propose a maximum-entropy relaxation of this problem. Multi-way cardinality constraints are matched in expectation rather than exactly, yielding an exponential-family distribution over complete population assignments and a convex optimization problem over Lagrange multipliers. We evaluate the approach on NPORS-derived scaling benchmarks with 4 to 40 attributes and compare it primarily against generalized raking. The results show that MaxEnt becomes increasingly advantageous as the number of attributes and ternary interactions grows, while raking remains competitive on smaller, lower-arity instances.
MetagenBERT: a Transformer-based Architecture using Foundational genomic Large Language Models for novel Metagenome Representation
Jan 05, 2026Metagenomic disease prediction commonly relies on species abundance tables derived from large, incomplete reference catalogs, constraining resolution and discarding valuable information contained in DNA reads. To overcome these limitations, we introduce MetagenBERT, a Transformer based framework that produces end to end metagenome embeddings directly from raw DNA sequences, without taxonomic or functional annotations. Reads are embedded using foundational genomic language models (DNABERT2 and the microbiome specialized DNABERTMS), then aggregated through a scalable clustering strategy based on FAISS accelerated KMeans. Each metagenome is represented as a cluster abundance vector summarizing the distribution of its embedded reads. We evaluate this approach on five benchmark gut microbiome datasets (Cirrhosis, T2D, Obesity, IBD, CRC). MetagenBERT achieves competitive or superior AUC performance relative to species abundance baselines across most tasks. Concatenating both representations further improves prediction, demonstrating complementarity between taxonomic and embedding derived signals. Clustering remains robust when applied to as little as 10% of reads, highlighting substantial redundancy in metagenomes and enabling major computational gains. We additionally introduce MetagenBERT Glob Mcardis, a cross cohort variant trained on the large, phenotypically diverse MetaCardis cohort and transferred to other datasets, retaining predictive signal including for unseen phenotypes, indicating the feasibility of a foundation model for metagenome representation. Robustness analyses (PERMANOVA, PERMDISP, entropy) show consistent separation of different states across subsamples. Overall, MetagenBERT provides a scalable, annotation free representation of metagenomes pointing toward future phenotype aware generalization across heterogeneous cohorts and sequencing technologies.
PlantSAM: An Object Detection-Driven Segmentation Pipeline for Herbarium Specimens
Jul 22, 2025Deep learning-based classification of herbarium images is hampered by background heterogeneity, which introduces noise and artifacts that can potentially mislead models and reduce classification accuracy. Addressing these background-related challenges is critical to improving model performance. We introduce PlantSAM, an automated segmentation pipeline that integrates YOLOv10 for plant region detection and the Segment Anything Model (SAM2) for segmentation. YOLOv10 generates bounding box prompts to guide SAM2, enhancing segmentation accuracy. Both models were fine-tuned on herbarium images and evaluated using Intersection over Union (IoU) and Dice coefficient metrics. PlantSAM achieved state-of-the-art segmentation performance, with an IoU of 0.94 and a Dice coefficient of 0.97. Incorporating segmented images into classification models led to consistent performance improvements across five tested botanical traits, with accuracy gains of up to 4.36% and F1-score improvements of 4.15%. Our findings highlight the importance of background removal in herbarium image analysis, as it significantly enhances classification accuracy by allowing models to focus more effectively on the foreground plant structures.
IKrNet: A Neural Network for Detecting Specific Drug-Induced Patterns in Electrocardiograms Amidst Physiological Variability
May 12, 2025Monitoring and analyzing electrocardiogram (ECG) signals, even under varying physiological conditions, including those influenced by physical activity, drugs and stress, is crucial to accurately assess cardiac health. However, current AI-based methods often fail to account for how these factors interact and alter ECG patterns, ultimately limiting their applicability in real-world settings. This study introduces IKrNet, a novel neural network model, which identifies drug-specific patterns in ECGs amidst certain physiological conditions. IKrNet's architecture incorporates spatial and temporal dynamics by using a convolutional backbone with varying receptive field size to capture spatial features. A bi-directional Long Short-Term Memory module is also employed to model temporal dependencies. By treating heart rate variability as a surrogate for physiological fluctuations, we evaluated IKrNet's performance across diverse scenarios, including conditions with physical stress, drug intake alone, and a baseline without drug presence. Our assessment follows a clinical protocol in which 990 healthy volunteers were administered 80mg of Sotalol, a drug which is known to be a precursor to Torsades-de-Pointes, a life-threatening arrhythmia. We show that IKrNet outperforms state-of-the-art models' accuracy and stability in varying physiological conditions, underscoring its clinical viability.
Prompting ChatGPT for Chinese Learning as L2: A CEFR and EBCL Level Study
Jan 25, 2025


The use of chatbots in language learning has evolved significantly since the 1960s, becoming more sophisticated platforms as generative AI emerged. These tools now simulate natural conversations, adapting to individual learners' needs, including those studying Chinese. Our study explores how learners can use specific prompts to engage Large Language Models (LLM) as personalized chatbots, aiming to target their language level based on the Common European Framework of Reference for Languages (CEFR) and the European Benchmarking Chinese Language (EBCL) project. Focusing on A1, A1+ and A2 levels, we examine the teaching of Chinese, which presents unique challenges due to its logographic writing system. Our goal is to develop prompts that integrate oral and written skills, using high-frequency character lists and controlling oral lexical productions. These tools, powered by generative AI, aim to enhance language practice by crossing lexical and sinographic recurrence. While generative AI shows potential as a personalized tutor, further evaluation is needed to assess its effectiveness. We conducted a systematic series of experiments using ChatGPT models to evaluate their adherence to constraints specified in the prompts. The results indicate that incorporating level A1 and A1+ characters, along with the associated reference list, significantly enhances compliance with the EBCL character set. Properly prompted, LLMs can increase exposure to the target language and offer interactive exchanges to develop language skills.
A text-to-tabular approach to generate synthetic patient data using LLMs
Dec 06, 2024



Access to large-scale high-quality healthcare databases is key to accelerate medical research and make insightful discoveries about diseases. However, access to such data is often limited by patient privacy concerns, data sharing restrictions and high costs. To overcome these limitations, synthetic patient data has emerged as an alternative. However, synthetic data generation (SDG) methods typically rely on machine learning (ML) models trained on original data, leading back to the data scarcity problem. We propose an approach to generate synthetic tabular patient data that does not require access to the original data, but only a description of the desired database. We leverage prior medical knowledge and in-context learning capabilities of large language models (LLMs) to generate realistic patient data, even in a low-resource setting. We quantitatively evaluate our approach against state-of-the-art SDG models, using fidelity, privacy, and utility metrics. Our results show that while LLMs may not match the performance of state-of-the-art models trained on the original data, they effectively generate realistic patient data with well-preserved clinical correlations. An ablation study highlights key elements of our prompt contributing to high-quality synthetic patient data generation. This approach, which is easy to use and does not require original data or advanced ML skills, is particularly valuable for quickly generating custom-designed patient data, supporting project implementation and providing educational resources.
Disease Classification in Metagenomics with 2D Embeddings and Deep Learning
Jun 23, 2018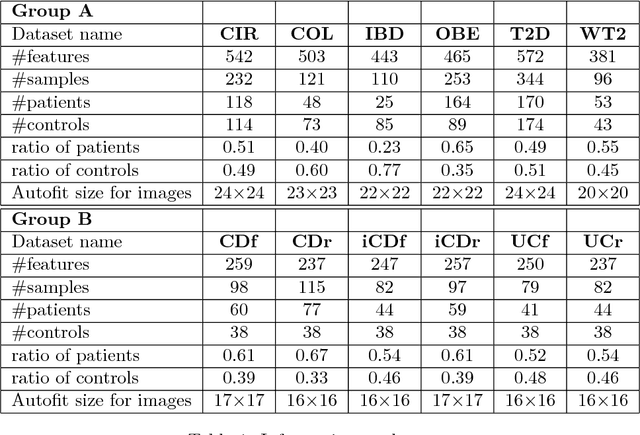
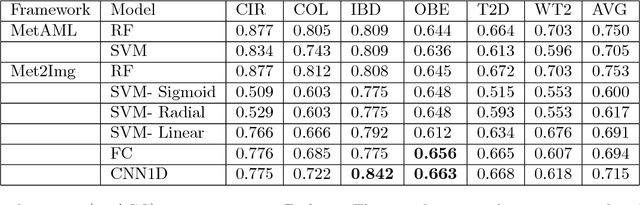
Deep learning (DL) techniques have shown unprecedented success when applied to images, waveforms, and text. Generally, when the sample size ($N$) is much bigger than the number of features ($d$), DL often outperforms other machine learning (ML) techniques, often through the use of Convolutional Neural Networks (CNNs). However, in many bioinformatics fields (including metagenomics), we encounter the opposite situation where $d$ is significantly greater than $N$. In these situations, applying DL techniques would lead to severe overfitting. Here we aim to improve classification of various diseases with metagenomic data through the use of CNNs. For this we proposed to represent metagenomic data as images. The proposed Met2Img approach relies on taxonomic and t-SNE embeddings to transform abundance data into "synthetic images". We applied our approach to twelve benchmark data sets including more than 1400 metagenomic samples. Our results show significant improvements over the state-of-the-art algorithms (Random Forest (RF), Support Vector Machine (SVM)). We observe that the integration of phylogenetic information alongside abundance data improves classification. The proposed approach is not only important in classification setting but also allows to visualize complex metagenomic data. The Met2Img is implemented in Python.
Deep Learning for Metagenomic Data: using 2D Embeddings and Convolutional Neural Networks
Dec 01, 2017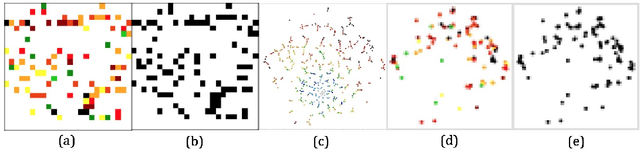
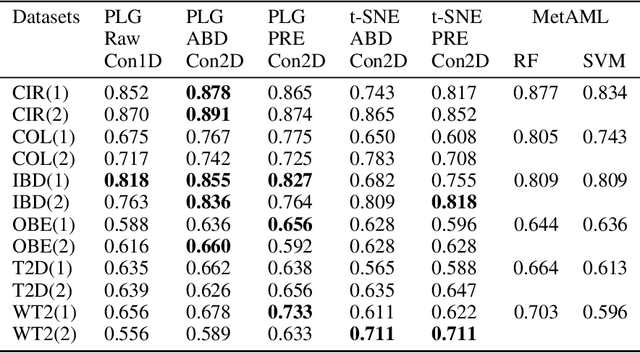
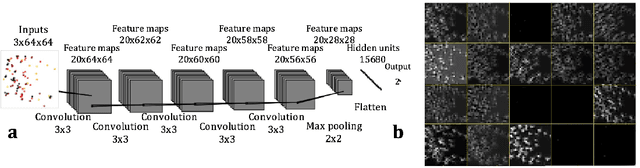
Deep learning (DL) techniques have had unprecedented success when applied to images, waveforms, and texts to cite a few. In general, when the sample size (N) is much greater than the number of features (d), DL outperforms previous machine learning (ML) techniques, often through the use of convolution neural networks (CNNs). However, in many bioinformatics ML tasks, we encounter the opposite situation where d is greater than N. In these situations, applying DL techniques (such as feed-forward networks) would lead to severe overfitting. Thus, sparse ML techniques (such as LASSO e.g.) usually yield the best results on these tasks. In this paper, we show how to apply CNNs on data which do not have originally an image structure (in particular on metagenomic data). Our first contribution is to show how to map metagenomic data in a meaningful way to 1D or 2D images. Based on this representation, we then apply a CNN, with the aim of predicting various diseases. The proposed approach is applied on six different datasets including in total over 1000 samples from various diseases. This approach could be a promising one for prediction tasks in the bioinformatics field.
Use of the C4.5 machine learning algorithm to test a clinical guideline-based decision support system
Dec 03, 2013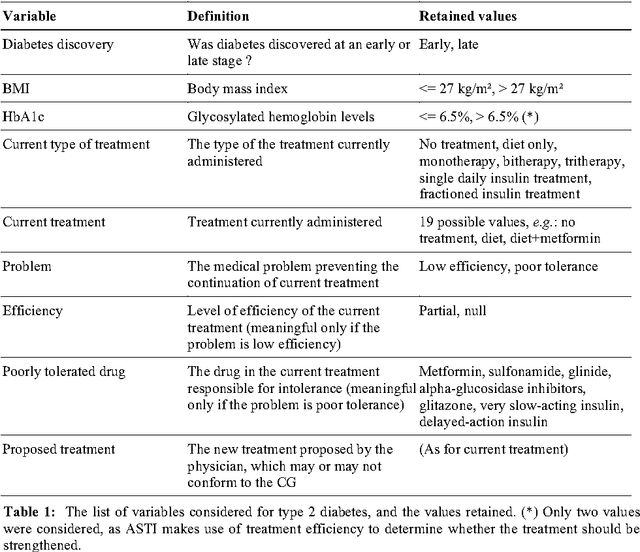

Well-designed medical decision support system (DSS) have been shown to improve health care quality. However, before they can be used in real clinical situations, these systems must be extensively tested, to ensure that they conform to the clinical guidelines (CG) on which they are based. Existing methods cannot be used for the systematic testing of all possible test cases. We describe here a new exhaustive dynamic verification method. In this method, the DSS is considered to be a black box, and the Quinlan C4.5 algorithm is used to build a decision tree from an exhaustive set of DSS input vectors and outputs. This method was successfully used for the testing of a medical DSS relating to chronic diseases: the ASTI critiquing module for type 2 diabetes.