 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSupervised deep learning prediction of the formation enthalpy of the full set of configurations in complex phases: the $σ-$phase as an example
Paper and Code
Nov 21, 2020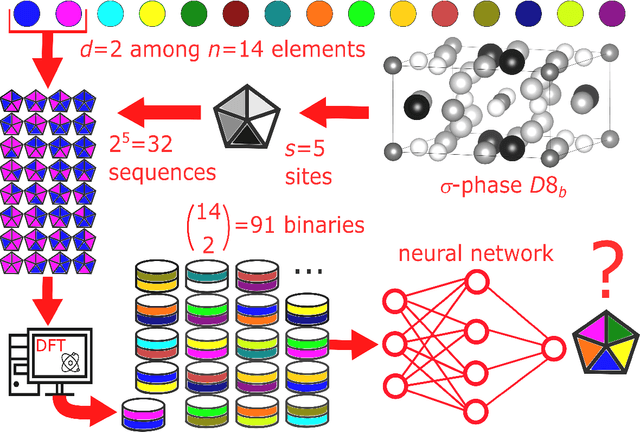
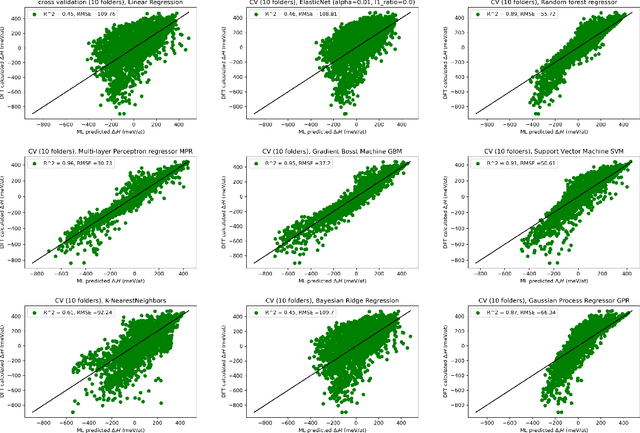
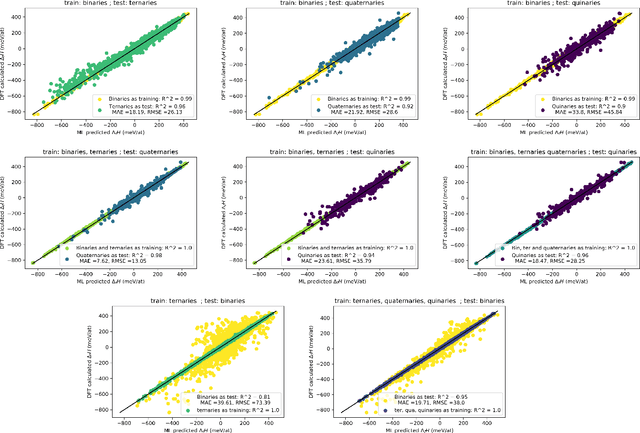
Machine learning (ML) methods are becoming integral to scientific inquiry in numerous disciplines, such as material sciences. In this manuscript, we demonstrate how ML can be used to predict several properties in solid-state chemistry, in particular the heat of formation of a given complex crystallographic phase (here the $\sigma-$phase, $tP30$, $D8_{b}$). Based on an independent and unprecedented large first principles dataset containing about 10,000 $\sigma-$compounds with $n=14$ different elements, we used a supervised learning approach, to predict all the $\sim$500,000 possible configurations within a mean absolute error of 23 meV/at ($\sim$2 kJ.mol$^{-1}$) on the heat of formation and $\sim$0.06 Ang. on the tetragonal cell parameters. We showed that neural network regression algorithms provide a significant improvement in accuracy of the predicted output compared to traditional regression techniques. Adding descriptors having physical nature (atomic radius, number of valence electrons) improves the learning precision. Based on our analysis, the training database composed of the only binary-compositions plays a major role in predicting the higher degree system configurations. Our result opens a broad avenue to efficient high-throughput investigations of the combinatorial binary calculation for multicomponent prediction of a complex phase.