 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA practical guide to machine learning interatomic potentials -- Status and future
Mar 12, 2025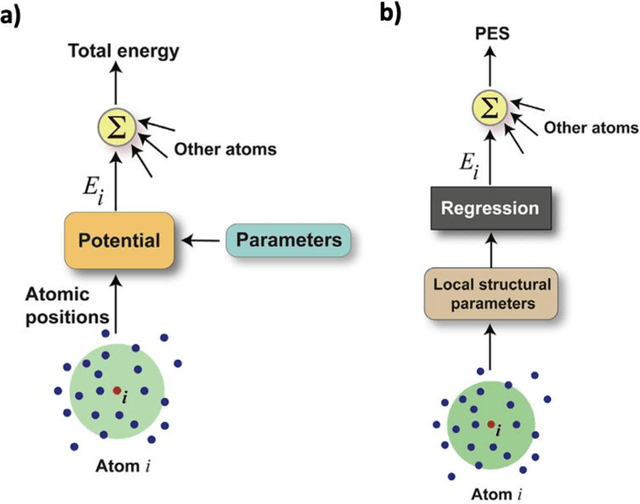

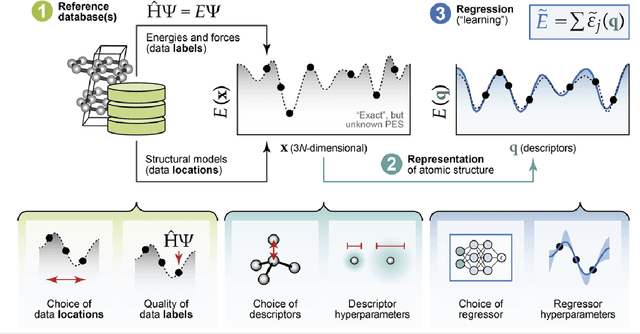
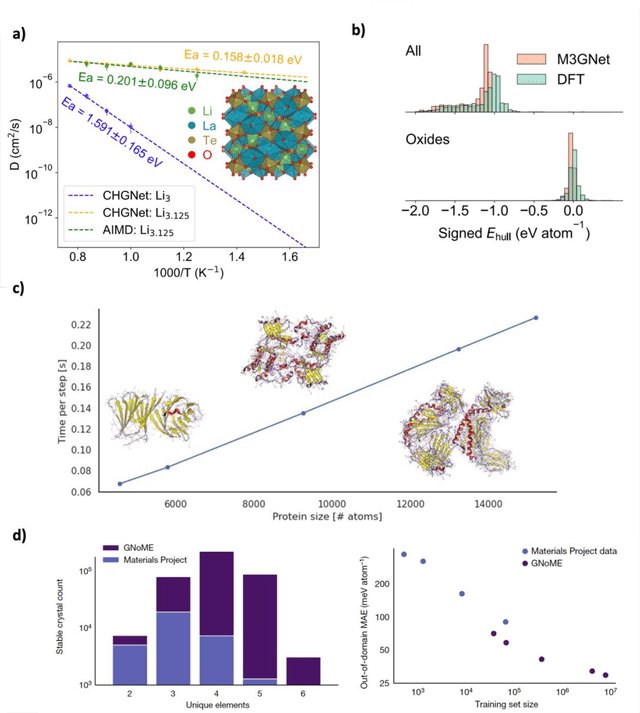
The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.
Excited-state nonadiabatic dynamics in explicit solvent using machine learned interatomic potentials
Jan 28, 2025Excited-state nonadiabatic simulations with quantum mechanics/molecular mechanics (QM/MM) are essential to understand photoinduced processes in explicit environments. However, the high computational cost of the underlying quantum chemical calculations limits its application in combination with trajectory surface hopping methods. Here, we use FieldSchNet, a machine-learned interatomic potential capable of incorporating electric field effects into the electronic states, to replace traditional QM/MM electrostatic embedding with its ML/MM counterpart for nonadiabatic excited state trajectories. The developed method is applied to furan in water, including five coupled singlet states. Our results demonstrate that with sufficiently curated training data, the ML/MM model reproduces the electronic kinetics and structural rearrangements of QM/MM surface hopping reference simulations. Furthermore, we identify performance metrics that provide robust and interpretable validation of model accuracy.
Deep Learning for UV Absorption Spectra with SchNarc: First Steps Towards Transferability in Chemical Compound Space
Jul 15, 2020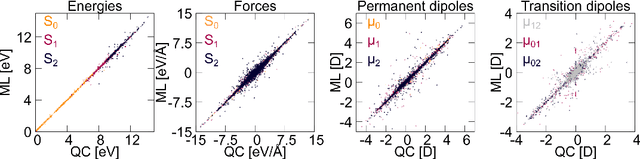
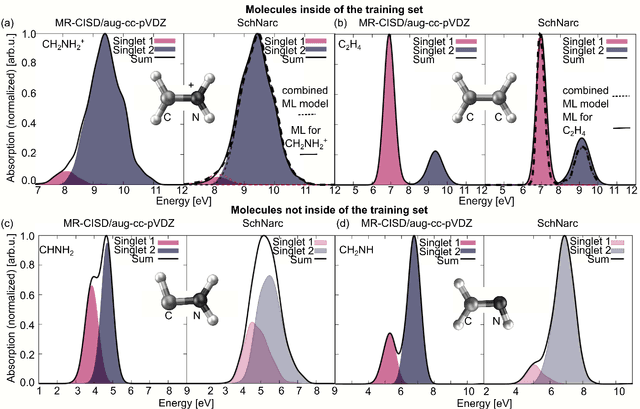
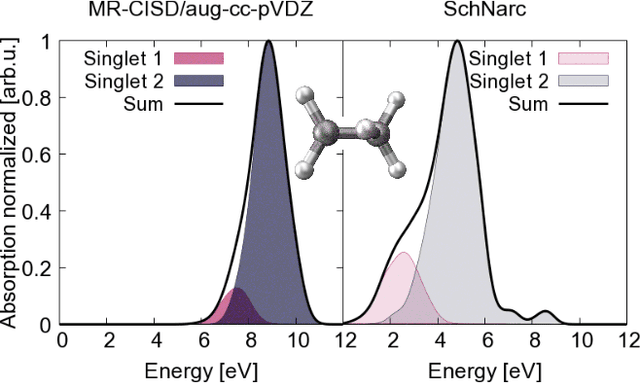

Machine learning (ML) has shown to advance the research field of quantum chemistry in almost any possible direction and has recently also entered the excited states to investigate the multifaceted photochemistry of molecules. In this paper, we pursue two goals: i) We show how ML can be used to model permanent dipole moments for excited states and transition dipole moments by adapting the charge model of [Chem. Sci., 2017, 8, 6924-6935], which was originally proposed for the permanent dipole moment vector of the electronic ground state. ii) We investigate the transferability of our excited-state ML models in chemical space, i.e., whether an ML model can predict properties of molecules that it has never been trained on and whether it can learn the different excited states of two molecules simultaneously. To this aim, we employ and extend our previously reported SchNarc approach for excited-state ML. We calculate UV absorption spectra from excited-state energies and transition dipole moments as well as electrostatic potentials from latent charges inferred by the ML model of the permanent dipole moment vectors. We train our ML models on CH$_2$NH$_2^+$ and C$_2$H$_4$, while predictions are carried out for these molecules and additionally for CHNH$_2$, CH$_2$NH, and C$_2$H$_5^+$. The results indicate that transferability is possible for the excited states.
Machine learning for electronically excited states of molecules
Jul 10, 2020


Electronically excited states of molecules are at the heart of photochemistry, photophysics, as well as photobiology and also play a role in material science. Their theoretical description requires highly accurate quantum chemical calculations, which are computationally expensive. In this review, we focus on how machine learning is employed not only to speed up such excited-state simulations but also how this branch of artificial intelligence can be used to advance this exciting research field in all its aspects. Discussed applications of machine learning for excited states include excited-state dynamics simulations, static calculations of absorption spectra, as well as many others. In order to put these studies into context, we discuss the promises and pitfalls of the involved machine learning techniques. Since the latter are mostly based on quantum chemistry calculations, we also provide a short introduction into excited-state electronic structure methods, approaches for nonadiabatic dynamics simulations and describe tricks and problems when using them in machine learning for excited states of molecules.
Machine learning and excited-state molecular dynamics
May 28, 2020

Machine learning is employed at an increasing rate in the research field of quantum chemistry. While the majority of approaches target the investigation of chemical systems in their electronic ground state, the inclusion of light into the processes leads to electronically excited states and gives rise to several new challenges. Here, we survey recent advances for excited-state dynamics based on machine learning. In doing so, we highlight successes, pitfalls, challenges and future avenues for machine learning approaches for light-induced molecular processes.
Combining SchNet and SHARC: The SchNarc machine learning approach for excited-state dynamics
Feb 17, 2020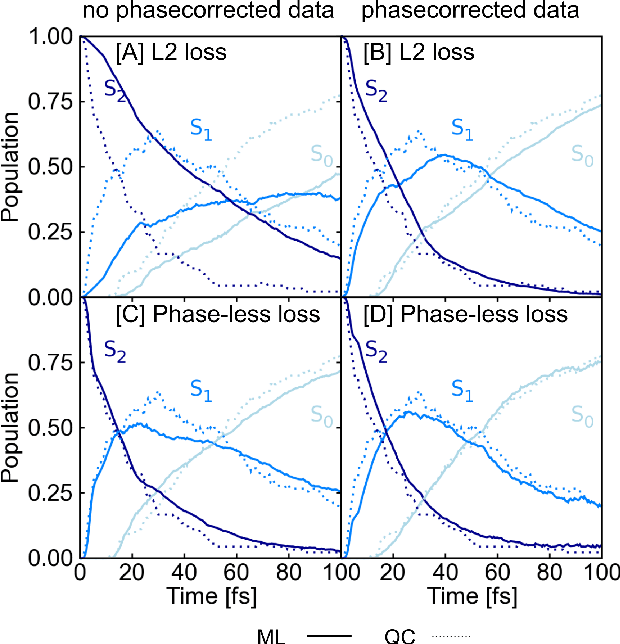
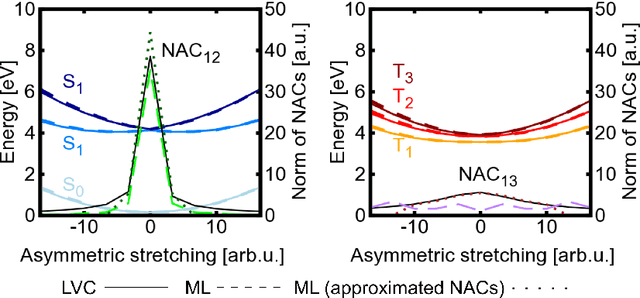
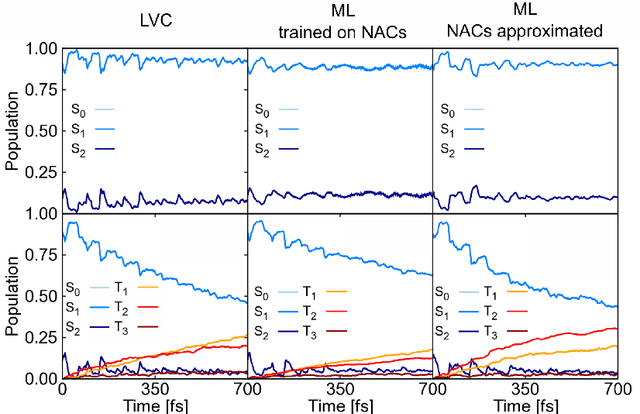
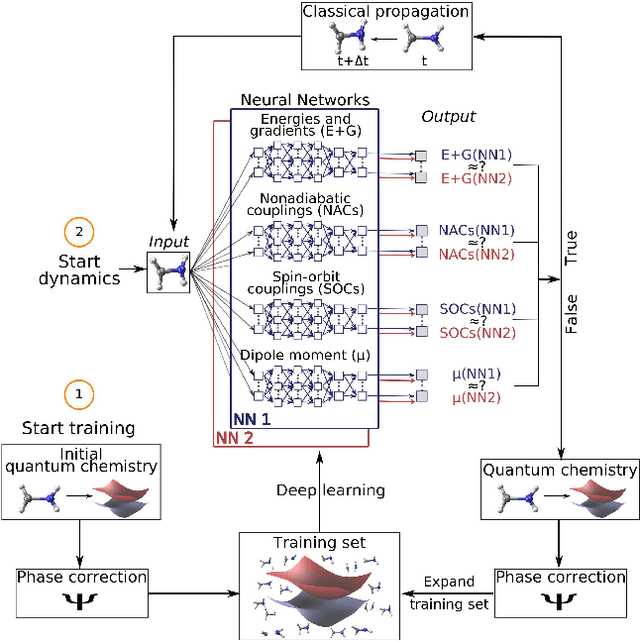
In recent years, deep learning has become a part of our everyday life and is revolutionizing quantum chemistry as well. In this work, we show how deep learning can be used to advance the research field of photochemistry by learning all important properties for photodynamics simulations. The properties are multiple energies, forces, nonadiabatic couplings and spin-orbit couplings. The nonadiabatic couplings are learned in a phase-free manner as derivatives of a virtually constructed property by the deep learning model, which guarantees rotational covariance. Additionally, an approximation for nonadiabatic couplings is introduced, based on the potentials, their gradients and Hessians. As deep-learning method, we employ SchNet extended for multiple electronic states. In combination with the molecular dynamics program SHARC, our approach termed SchNarc is tested on a model system and two realistic polyatomic molecules and paves the way towards efficient photodynamics simulations of complex systems.
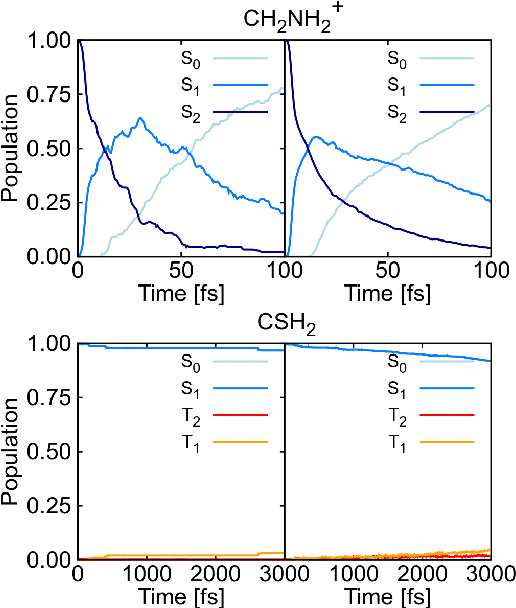
Neural networks and kernel ridge regression for excited states dynamics of CH$_2$NH$_2^+$: From single-state to multi-state representations and multi-property machine learning models
Dec 18, 2019

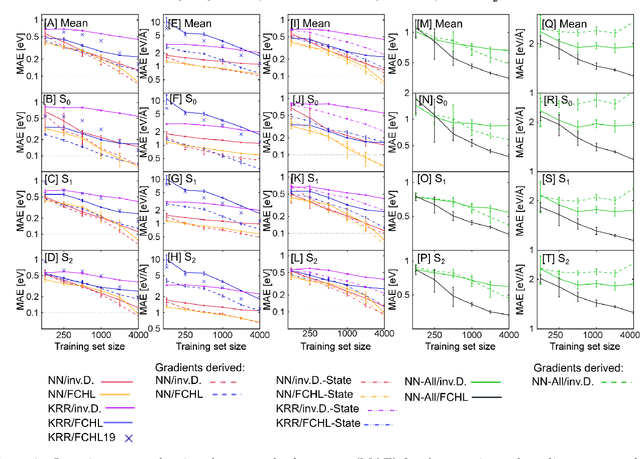

Excited-state dynamics simulations are a powerful tool to investigate photo-induced reactions of molecules and materials and provide complementary information to experiments. Since the applicability of these simulation techniques is limited by the costs of the underlying electronic structure calculations, we develop and assess different machine learning models for this task. The machine learning models are trained on {\emph ab initio} calculations for excited electronic states, using the methylenimmonium cation (CH$_2$NH$_2^+$) as a model system. For the prediction of excited-state properties, multiple outputs are desirable, which is straightforward with neural networks but less explored with kernel ridge regression. We overcome this challenge for kernel ridge regression in the case of energy predictions by encoding the electronic states explicitly in the inputs, in addition to the molecular representation. We adopt this strategy also for our neural networks for comparison. Such a state encoding enables not only kernel ridge regression with multiple outputs but leads also to more accurate machine learning models for state-specific properties. An important goal for excited-state machine learning models is their use in dynamics simulations, which needs not only state-specific information but also couplings, i.e., properties involving pairs of states. Accordingly, we investigate the performance of different models for such coupling elements. Furthermore, we explore how combining all properties in a single neural network affects the accuracy. As an ultimate test for our machine learning models, we carry out excited-state dynamics simulations based on the predicted energies, forces and couplings and, thus, show the scopes and possibilities of machine learning for the treatment of electronically excited states.
Machine learning enables long time scale molecular photodynamics simulations
Nov 22, 2018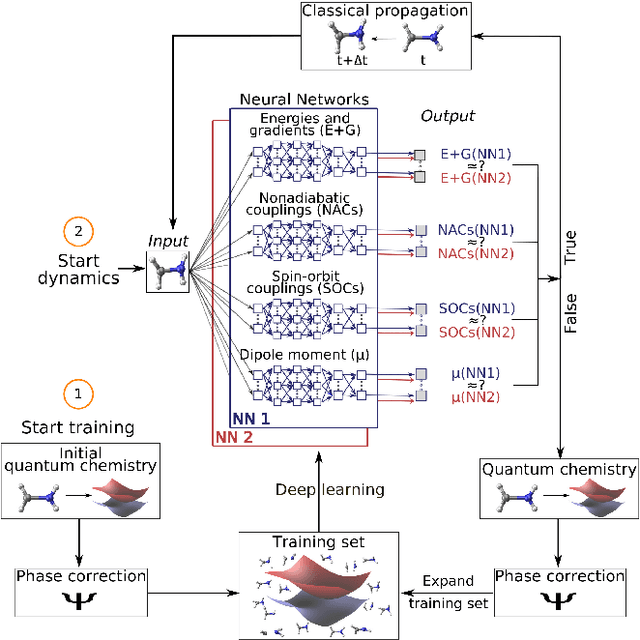
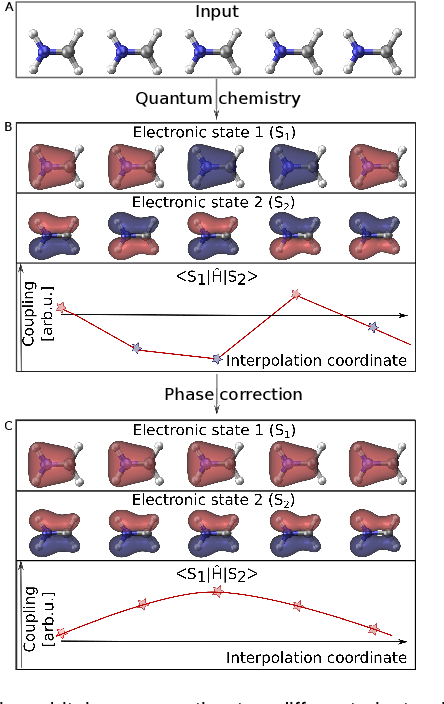
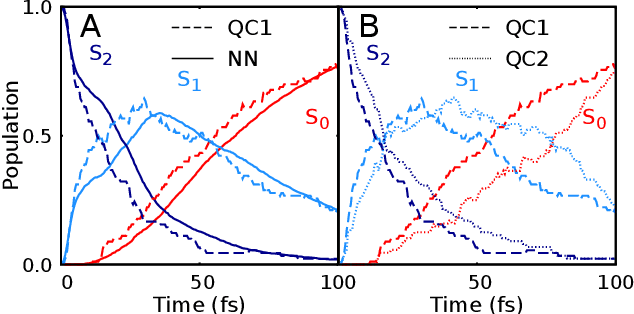
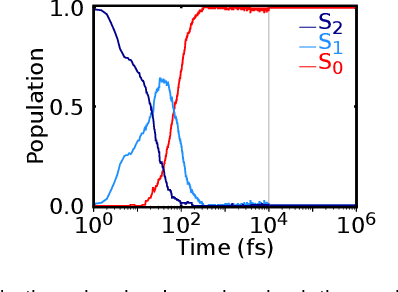
Photo-induced processes are fundamental in nature, but accurate simulations are seriously limited by the cost of the underlying quantum chemical calculations, hampering their application for long time scales. Here we introduce a method based on machine learning to overcome this bottleneck and enable accurate photodynamics on nanosecond time scales, which are otherwise out of reach with contemporary approaches. Instead of expensive quantum chemistry during molecular dynamics simulations, we use deep neural networks to learn the relationship between a molecular geometry and its high-dimensional electronic properties. As an example, the time evolution of the methylenimmonium cation for one nanosecond is used to demonstrate that machine learning algorithms can outperform standard excited-state molecular dynamics approaches in their computational efficiency while delivering the same accuracy.