 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHOAX: A Hyperparameter Optimization Algorithm Explorer for Neural Networks
Feb 01, 2023Computational chemistry has become an important tool to predict and understand molecular properties and reactions. Even though recent years have seen a significant growth in new algorithms and computational methods that speed up quantum chemical calculations, the bottleneck for trajectory-based methods to study photoinduced processes is still the huge number of electronic structure calculations. In this work, we present an innovative solution, in which the amount of electronic structure calculations is drastically reduced, by employing machine learning algorithms and methods borrowed from the realm of artificial intelligence. However, applying these algorithms effectively requires finding optimal hyperparameters, which remains a challenge itself. Here we present an automated user-friendly framework, HOAX, to perform the hyperparameter optimization for neural networks, which bypasses the need for a lengthy manual process. The neural network generated potential energy surfaces (PESs) reduces the computational costs compared to the ab initio-based PESs. We perform a comparative investigation on the performance of different hyperparameter optimiziation algorithms, namely grid search, simulated annealing, genetic algorithm, and bayesian optimizer in finding the optimal hyperparameters necessary for constructing the well-performing neural network in order to fit the PESs of small organic molecules. Our results show that this automated toolkit not only facilitate a straightforward way to perform the hyperparameter optimization but also the resulting neural networks-based generated PESs are in reasonable agreement with the ab initio-based PESs.
Machine learning enables long time scale molecular photodynamics simulations
Nov 22, 2018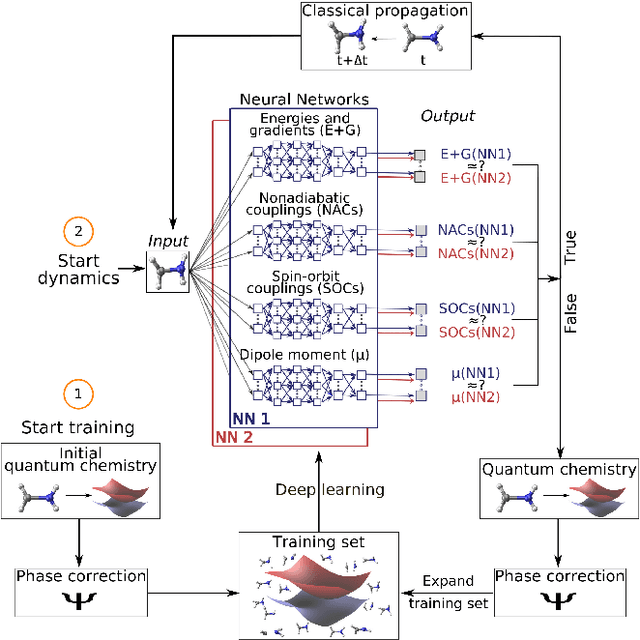
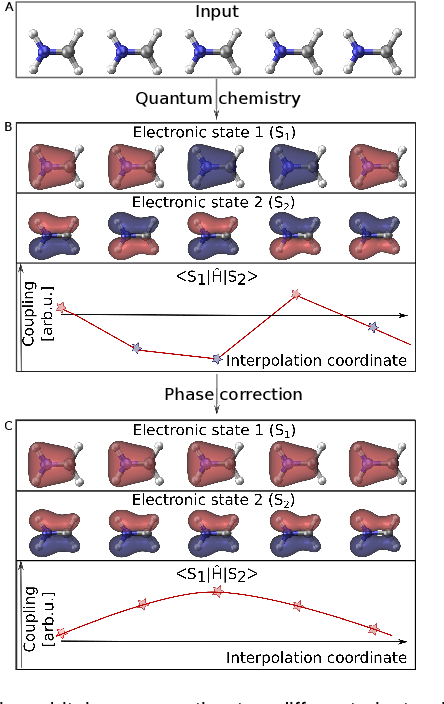
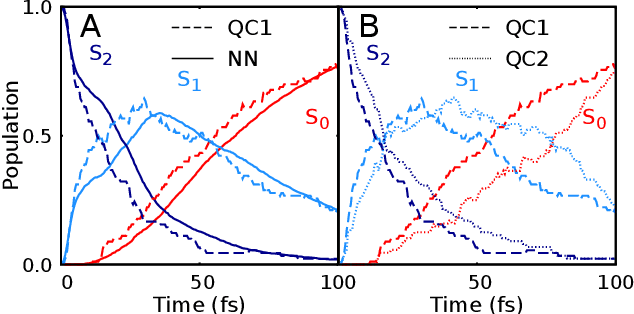
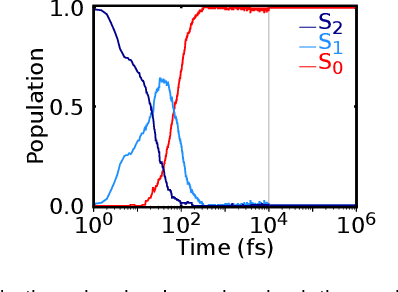
Photo-induced processes are fundamental in nature, but accurate simulations are seriously limited by the cost of the underlying quantum chemical calculations, hampering their application for long time scales. Here we introduce a method based on machine learning to overcome this bottleneck and enable accurate photodynamics on nanosecond time scales, which are otherwise out of reach with contemporary approaches. Instead of expensive quantum chemistry during molecular dynamics simulations, we use deep neural networks to learn the relationship between a molecular geometry and its high-dimensional electronic properties. As an example, the time evolution of the methylenimmonium cation for one nanosecond is used to demonstrate that machine learning algorithms can outperform standard excited-state molecular dynamics approaches in their computational efficiency while delivering the same accuracy.