 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExcited-state nonadiabatic dynamics in explicit solvent using machine learned interatomic potentials
Jan 28, 2025Excited-state nonadiabatic simulations with quantum mechanics/molecular mechanics (QM/MM) are essential to understand photoinduced processes in explicit environments. However, the high computational cost of the underlying quantum chemical calculations limits its application in combination with trajectory surface hopping methods. Here, we use FieldSchNet, a machine-learned interatomic potential capable of incorporating electric field effects into the electronic states, to replace traditional QM/MM electrostatic embedding with its ML/MM counterpart for nonadiabatic excited state trajectories. The developed method is applied to furan in water, including five coupled singlet states. Our results demonstrate that with sufficiently curated training data, the ML/MM model reproduces the electronic kinetics and structural rearrangements of QM/MM surface hopping reference simulations. Furthermore, we identify performance metrics that provide robust and interpretable validation of model accuracy.
Machine learning enables long time scale molecular photodynamics simulations
Nov 22, 2018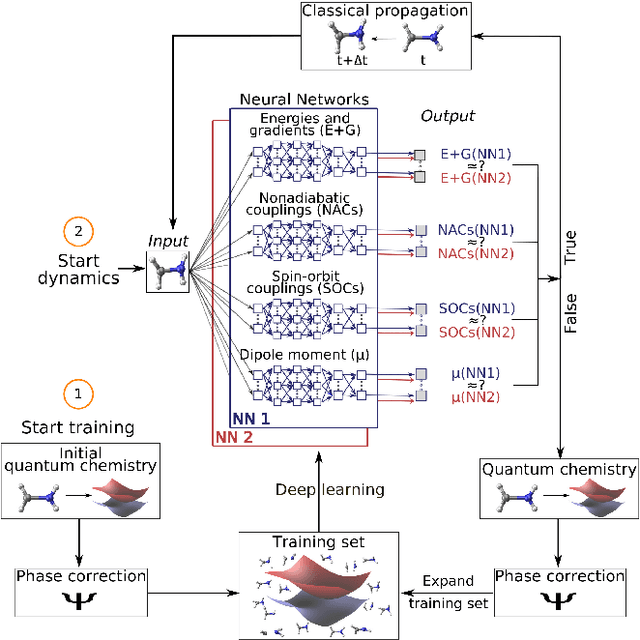
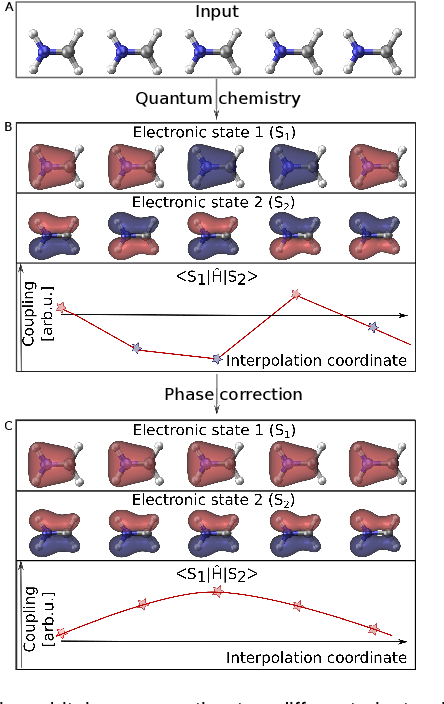
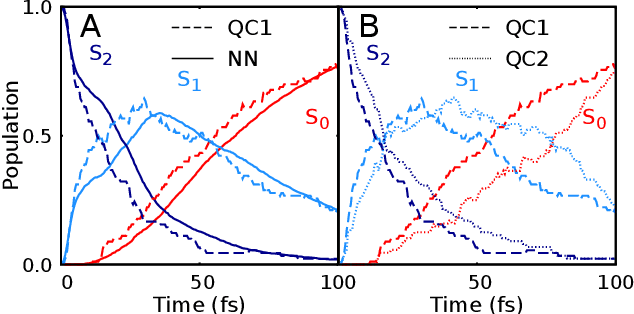
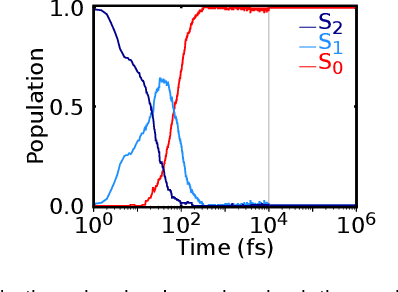
Photo-induced processes are fundamental in nature, but accurate simulations are seriously limited by the cost of the underlying quantum chemical calculations, hampering their application for long time scales. Here we introduce a method based on machine learning to overcome this bottleneck and enable accurate photodynamics on nanosecond time scales, which are otherwise out of reach with contemporary approaches. Instead of expensive quantum chemistry during molecular dynamics simulations, we use deep neural networks to learn the relationship between a molecular geometry and its high-dimensional electronic properties. As an example, the time evolution of the methylenimmonium cation for one nanosecond is used to demonstrate that machine learning algorithms can outperform standard excited-state molecular dynamics approaches in their computational efficiency while delivering the same accuracy.