 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCHIMERA-Bench: A Benchmark Dataset for Epitope-Specific Antibody Design
Mar 13, 2026Computational antibody design has seen rapid methodological progress, with dozens of deep generative methods proposed in the past three years, yet the field lacks a standardized benchmark for fair comparison and model development. These methods are evaluated on different SAbDab snapshots, non-overlapping test sets, and incompatible metrics, and the literature fragments the design problem into numerous sub-tasks with no common definition. We introduce \textsc{Chimera-Bench} (\textbf{C}DR \textbf{M}odeling with \textbf{E}pitope-guided \textbf{R}edesign), a unified benchmark built around a single canonical task: \emph{epitope-conditioned CDR sequence-structure co-design}. \textsc{Chimera-Bench} provides (1) a curated, deduplicated dataset of \textbf{2,922} antibody-antigen complexes with epitope and paratope annotations; (2) three biologically motivated splits testing generalization to unseen epitopes, unseen antigen folds, and prospective temporal targets; and (3) a comprehensive evaluation protocol with five metric groups including novel epitope-specificity measures. We benchmark representative methods spanning different generative paradigms and report results across all splits. \textsc{Chimera-Bench} is the largest dataset of its kind for the antibody design problem, allowing the community to develop and test novel methods and evaluate their generalizability. The source code and data are available at: https://github.com/mansoor181/chimera-bench.git
Converting Time Series Data to Numeric Representations Using Alphabetic Mapping and k-mer strategy
Dec 29, 2024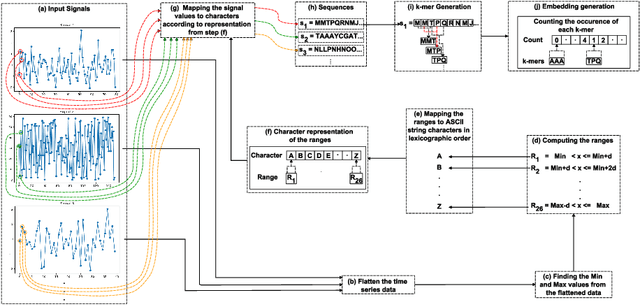
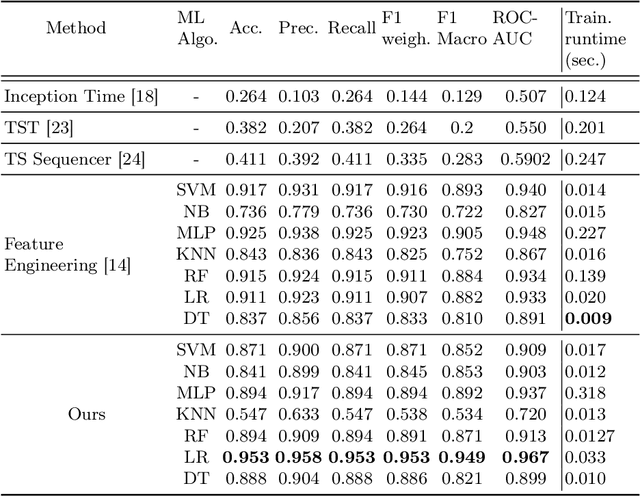
In the realm of data analysis and bioinformatics, representing time series data in a manner akin to biological sequences offers a novel approach to leverage sequence analysis techniques. Transforming time series signals into molecular sequence-type representations allows us to enhance pattern recognition by applying sophisticated sequence analysis techniques (e.g. $k$-mers based representation) developed in bioinformatics, uncovering hidden patterns and relationships in complex, non-linear time series data. This paper proposes a method to transform time series signals into biological/molecular sequence-type representations using a unique alphabetic mapping technique. By generating 26 ranges corresponding to the 26 letters of the English alphabet, each value within the time series is mapped to a specific character based on its range. This conversion facilitates the application of sequence analysis algorithms, typically used in bioinformatics, to analyze time series data. We demonstrate the effectiveness of this approach by converting real-world time series signals into character sequences and performing sequence classification. The resulting sequences can be utilized for various sequence-based analysis techniques, offering a new perspective on time series data representation and analysis.
Hilbert Curve Based Molecular Sequence Analysis
Dec 29, 2024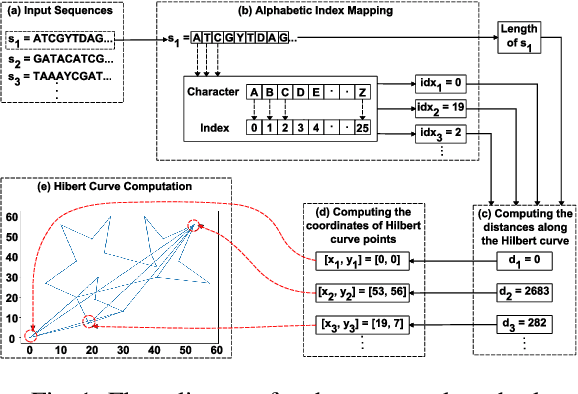
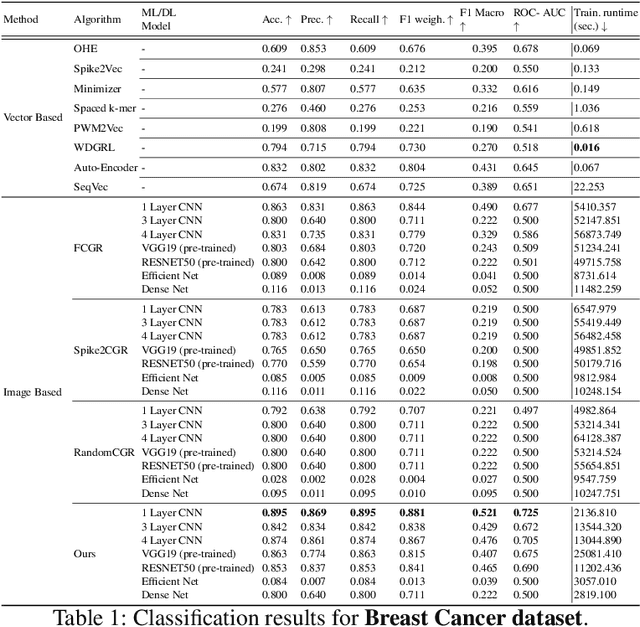
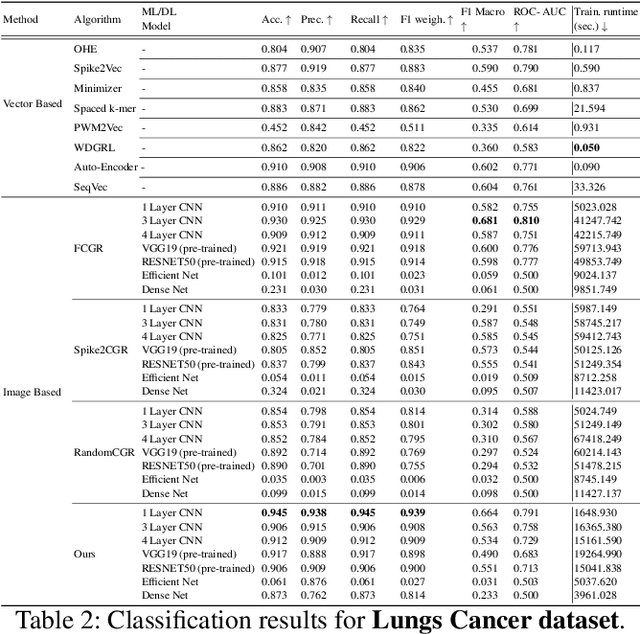
Accurate molecular sequence analysis is a key task in the field of bioinformatics. To apply molecular sequence classification algorithms, we first need to generate the appropriate representations of the sequences. Traditional numeric sequence representation techniques are mostly based on sequence alignment that faces limitations in the form of lack of accuracy. Although several alignment-free techniques have also been introduced, their tabular data form results in low performance when used with Deep Learning (DL) models compared to the competitive performance observed in the case of image-based data. To find a solution to this problem and to make Deep Learning (DL) models function to their maximum potential while capturing the important spatial information in the sequence data, we propose a universal Hibert curve-based Chaos Game Representation (CGR) method. This method is a transformative function that involves a novel Alphabetic index mapping technique used in constructing Hilbert curve-based image representation from molecular sequences. Our method can be globally applied to any type of molecular sequence data. The Hilbert curve-based image representations can be used as input to sophisticated vision DL models for sequence classification. The proposed method shows promising results as it outperforms current state-of-the-art methods by achieving a high accuracy of $94.5$\% and an F1 score of $93.9\%$ when tested with the CNN model on the lung cancer dataset. This approach opens up a new horizon for exploring molecular sequence analysis using image classification methods.
Computing Gram Matrix for SMILES Strings using RDKFingerprint and Sinkhorn-Knopp Algorithm
Dec 19, 2024In molecular structure data, SMILES (Simplified Molecular Input Line Entry System) strings are used to analyze molecular structure design. Numerical feature representation of SMILES strings is a challenging task. This work proposes a kernel-based approach for encoding and analyzing molecular structures from SMILES strings. The proposed approach involves computing a kernel matrix using the Sinkhorn-Knopp algorithm while using kernel principal component analysis (PCA) for dimensionality reduction. The resulting low-dimensional embeddings are then used for classification and regression analysis. The kernel matrix is computed by converting the SMILES strings into molecular structures using the Morgan Fingerprint, which computes a fingerprint for each molecule. The distance matrix is computed using the pairwise kernels function. The Sinkhorn-Knopp algorithm is used to compute the final kernel matrix that satisfies the constraints of a probability distribution. This is achieved by iteratively adjusting the kernel matrix until the marginal distributions of the rows and columns match the desired marginal distributions. We provided a comprehensive empirical analysis of the proposed kernel method to evaluate its goodness with greater depth. The suggested method is assessed for drug subcategory prediction (classification task) and solubility AlogPS ``Aqueous solubility and Octanol/Water partition coefficient" (regression task) using the benchmark SMILES string dataset. The outcomes show the proposed method outperforms several baseline methods in terms of supervised analysis and has potential uses in molecular design and drug discovery. Overall, the suggested method is a promising avenue for kernel methods-based molecular structure analysis and design.
Position Specific Scoring Is All You Need? Revisiting Protein Sequence Classification Tasks
Oct 16, 2024


Understanding the structural and functional characteristics of proteins are crucial for developing preventative and curative strategies that impact fields from drug discovery to policy development. An important and popular technique for examining how amino acids make up these characteristics of the protein sequences with position-specific scoring (PSS). While the string kernel is crucial in natural language processing (NLP), it is unclear if string kernels can extract biologically meaningful information from protein sequences, despite the fact that they have been shown to be effective in the general sequence analysis tasks. In this work, we propose a weighted PSS kernel matrix (or W-PSSKM), that combines a PSS representation of protein sequences, which encodes the frequency information of each amino acid in a sequence, with the notion of the string kernel. This results in a novel kernel function that outperforms many other approaches for protein sequence classification. We perform extensive experimentation to evaluate the proposed method. Our findings demonstrate that the W-PSSKM significantly outperforms existing baselines and state-of-the-art methods and achieves up to 45.1\% improvement in classification accuracy.
Sequence-Based Nanobody-Antigen Binding Prediction
Jul 15, 2023



Nanobodies (Nb) are monomeric heavy-chain fragments derived from heavy-chain only antibodies naturally found in Camelids and Sharks. Their considerably small size (~3-4 nm; 13 kDa) and favorable biophysical properties make them attractive targets for recombinant production. Furthermore, their unique ability to bind selectively to specific antigens, such as toxins, chemicals, bacteria, and viruses, makes them powerful tools in cell biology, structural biology, medical diagnostics, and future therapeutic agents in treating cancer and other serious illnesses. However, a critical challenge in nanobodies production is the unavailability of nanobodies for a majority of antigens. Although some computational methods have been proposed to screen potential nanobodies for given target antigens, their practical application is highly restricted due to their reliance on 3D structures. Moreover, predicting nanobodyantigen interactions (binding) is a time-consuming and labor-intensive task. This study aims to develop a machine-learning method to predict Nanobody-Antigen binding solely based on the sequence data. We curated a comprehensive dataset of Nanobody-Antigen binding and nonbinding data and devised an embedding method based on gapped k-mers to predict binding based only on sequences of nanobody and antigen. Our approach achieves up to 90% accuracy in binding prediction and is significantly more efficient compared to the widely-used computational docking technique.
CAMP: A Context-Aware Cricket Players Performance Metric
Jul 14, 2023Cricket is the second most popular sport after soccer in terms of viewership. However, the assessment of individual player performance, a fundamental task in team sports, is currently primarily based on aggregate performance statistics, including average runs and wickets taken. We propose Context-Aware Metric of player Performance, CAMP, to quantify individual players' contributions toward a cricket match outcome. CAMP employs data mining methods and enables effective data-driven decision-making for selection and drafting, coaching and training, team line-ups, and strategy development. CAMP incorporates the exact context of performance, such as opponents' strengths and specific circumstances of games, such as pressure situations. We empirically evaluate CAMP on data of limited-over cricket matches between 2001 and 2019. In every match, a committee of experts declares one player as the best player, called Man of the M}atch (MoM). The top two rated players by CAMP match with MoM in 83\% of the 961 games. Thus, the CAMP rating of the best player closely matches that of the domain experts. By this measure, CAMP significantly outperforms the current best-known players' contribution measure based on the Duckworth-Lewis-Stern (DLS) method.
Robust Brain Age Estimation via Regression Models and MRI-derived Features
Jun 08, 2023The determination of biological brain age is a crucial biomarker in the assessment of neurological disorders and understanding of the morphological changes that occur during aging. Various machine learning models have been proposed for estimating brain age through Magnetic Resonance Imaging (MRI) of healthy controls. However, developing a robust brain age estimation (BAE) framework has been challenging due to the selection of appropriate MRI-derived features and the high cost of MRI acquisition. In this study, we present a novel BAE framework using the Open Big Healthy Brain (OpenBHB) dataset, which is a new multi-site and publicly available benchmark dataset that includes region-wise feature metrics derived from T1-weighted (T1-w) brain MRI scans of 3965 healthy controls aged between 6 to 86 years. Our approach integrates three different MRI-derived region-wise features and different regression models, resulting in a highly accurate brain age estimation with a Mean Absolute Error (MAE) of 3.25 years, demonstrating the framework's robustness. We also analyze our model's regression-based performance on gender-wise (male and female) healthy test groups. The proposed BAE framework provides a new approach for estimating brain age, which has important implications for the understanding of neurological disorders and age-related brain changes.
Virus2Vec: Viral Sequence Classification Using Machine Learning
Apr 24, 2023Understanding the host-specificity of different families of viruses sheds light on the origin of, e.g., SARS-CoV-2, rabies, and other such zoonotic pathogens in humans. It enables epidemiologists, medical professionals, and policymakers to curb existing epidemics and prevent future ones promptly. In the family Coronaviridae (of which SARS-CoV-2 is a member), it is well-known that the spike protein is the point of contact between the virus and the host cell membrane. On the other hand, the two traditional mammalian orders, Carnivora (carnivores) and Chiroptera (bats) are recognized to be responsible for maintaining and spreading the Rabies Lyssavirus (RABV). We propose Virus2Vec, a feature-vector representation for viral (nucleotide or amino acid) sequences that enable vector-space-based machine learning models to identify viral hosts. Virus2Vec generates numerical feature vectors for unaligned sequences, allowing us to forego the computationally expensive sequence alignment step from the pipeline. Virus2Vec leverages the power of both the \emph{minimizer} and position weight matrix (PWM) to generate compact feature vectors. Using several classifiers, we empirically evaluate Virus2Vec on real-world spike sequences of Coronaviridae and rabies virus sequence data to predict the host (identifying the reservoirs of infection). Our results demonstrate that Virus2Vec outperforms the predictive accuracies of baseline and state-of-the-art methods.
BioSequence2Vec: Efficient Embedding Generation For Biological Sequences
Apr 01, 2023


Representation learning is an important step in the machine learning pipeline. Given the current biological sequencing data volume, learning an explicit representation is prohibitive due to the dimensionality of the resulting feature vectors. Kernel-based methods, e.g., SVM, are a proven efficient and useful alternative for several machine learning (ML) tasks such as sequence classification. Three challenges with kernel methods are (i) the computation time, (ii) the memory usage (storing an $n\times n$ matrix), and (iii) the usage of kernel matrices limited to kernel-based ML methods (difficult to generalize on non-kernel classifiers). While (i) can be solved using approximate methods, challenge (ii) remains for typical kernel methods. Similarly, although non-kernel-based ML methods can be applied to kernel matrices by extracting principal components (kernel PCA), it may result in information loss, while being computationally expensive. In this paper, we propose a general-purpose representation learning approach that embodies kernel methods' qualities while avoiding computation, memory, and generalizability challenges. This involves computing a low-dimensional embedding of each sequence, using random projections of its $k$-mer frequency vectors, significantly reducing the computation needed to compute the dot product and the memory needed to store the resulting representation. Our proposed fast and alignment-free embedding method can be used as input to any distance (e.g., $k$ nearest neighbors) and non-distance (e.g., decision tree) based ML method for classification and clustering tasks. Using different forms of biological sequences as input, we perform a variety of real-world classification tasks, such as SARS-CoV-2 lineage and gene family classification, outperforming several state-of-the-art embedding and kernel methods in predictive performance.