 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVirus2Vec: Viral Sequence Classification Using Machine Learning
Apr 24, 2023Understanding the host-specificity of different families of viruses sheds light on the origin of, e.g., SARS-CoV-2, rabies, and other such zoonotic pathogens in humans. It enables epidemiologists, medical professionals, and policymakers to curb existing epidemics and prevent future ones promptly. In the family Coronaviridae (of which SARS-CoV-2 is a member), it is well-known that the spike protein is the point of contact between the virus and the host cell membrane. On the other hand, the two traditional mammalian orders, Carnivora (carnivores) and Chiroptera (bats) are recognized to be responsible for maintaining and spreading the Rabies Lyssavirus (RABV). We propose Virus2Vec, a feature-vector representation for viral (nucleotide or amino acid) sequences that enable vector-space-based machine learning models to identify viral hosts. Virus2Vec generates numerical feature vectors for unaligned sequences, allowing us to forego the computationally expensive sequence alignment step from the pipeline. Virus2Vec leverages the power of both the \emph{minimizer} and position weight matrix (PWM) to generate compact feature vectors. Using several classifiers, we empirically evaluate Virus2Vec on real-world spike sequences of Coronaviridae and rabies virus sequence data to predict the host (identifying the reservoirs of infection). Our results demonstrate that Virus2Vec outperforms the predictive accuracies of baseline and state-of-the-art methods.
PWM2Vec: An Efficient Embedding Approach for Viral Host Specification from Coronavirus Spike Sequences
Jan 06, 2022
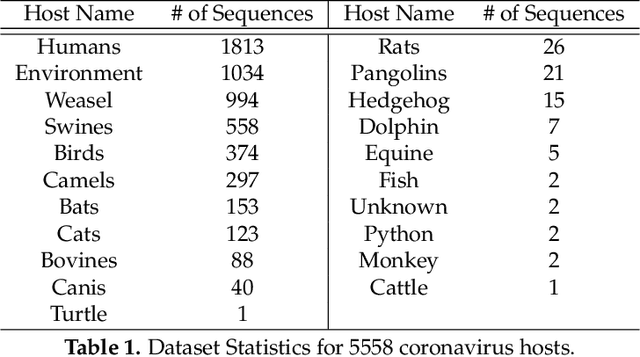
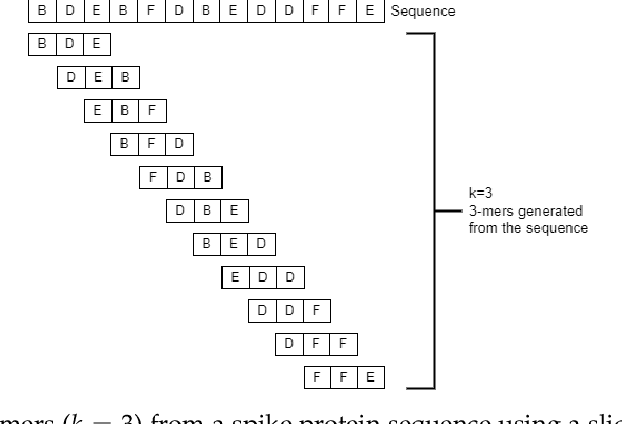
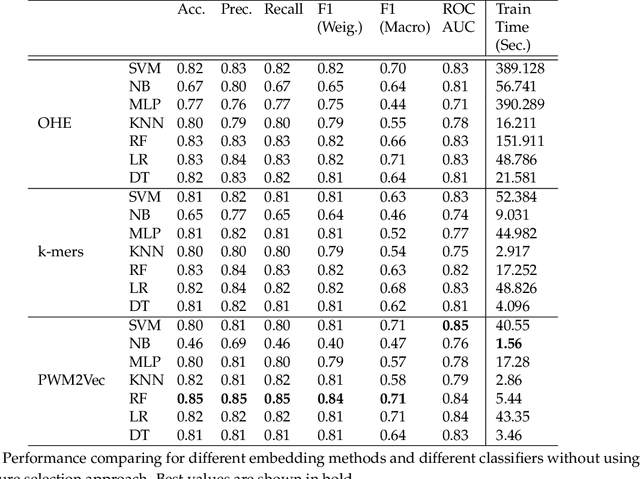
COVID-19 pandemic, is still unknown and is an important open question. There are speculations that bats are a possible origin. Likewise, there are many closely related (corona-) viruses, such as SARS, which was found to be transmitted through civets. The study of the different hosts which can be potential carriers and transmitters of deadly viruses to humans is crucial to understanding, mitigating and preventing current and future pandemics. In coronaviruses, the surface (S) protein, or spike protein, is an important part of determining host specificity since it is the point of contact between the virus and the host cell membrane. In this paper, we classify the hosts of over five thousand coronaviruses from their spike protein sequences, segregating them into clusters of distinct hosts among avians, bats, camels, swines, humans and weasels, to name a few. We propose a feature embedding based on the well-known position-weight matrix (PWM), which we call PWM2Vec, and use to generate feature vectors from the spike protein sequences of these coronaviruses. While our embedding is inspired by the success of PWMs in biological applications such as determining protein function, or identifying transcription factor binding sites, we are the first (to the best of our knowledge) to use PWMs in the context of host classification from viral sequences to generate a fixed-length feature vector representation. The results on the real world data show that in using PWM2Vec, we are able to perform comparably well as compared to baseline models. We also measure the importance of different amino acids using information gain to show the amino acids which are important for predicting the host of a given coronavirus.