 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePWM2Vec: An Efficient Embedding Approach for Viral Host Specification from Coronavirus Spike Sequences
Jan 06, 2022
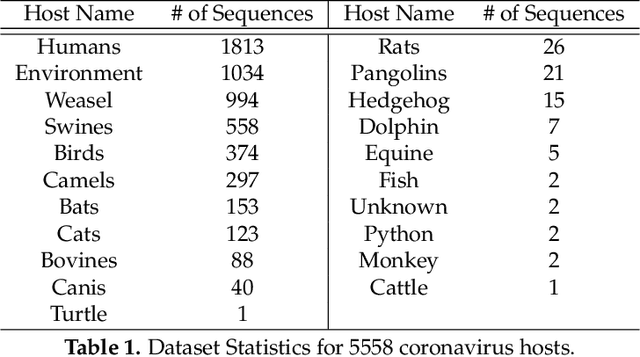
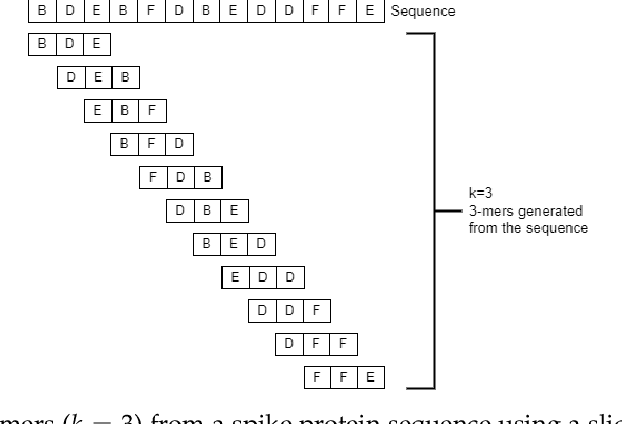
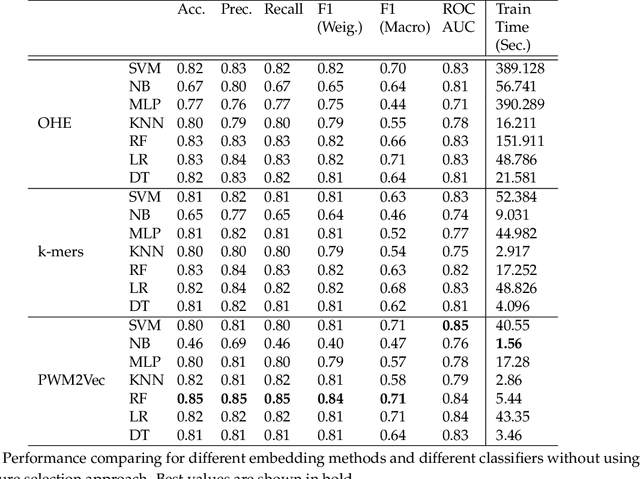
COVID-19 pandemic, is still unknown and is an important open question. There are speculations that bats are a possible origin. Likewise, there are many closely related (corona-) viruses, such as SARS, which was found to be transmitted through civets. The study of the different hosts which can be potential carriers and transmitters of deadly viruses to humans is crucial to understanding, mitigating and preventing current and future pandemics. In coronaviruses, the surface (S) protein, or spike protein, is an important part of determining host specificity since it is the point of contact between the virus and the host cell membrane. In this paper, we classify the hosts of over five thousand coronaviruses from their spike protein sequences, segregating them into clusters of distinct hosts among avians, bats, camels, swines, humans and weasels, to name a few. We propose a feature embedding based on the well-known position-weight matrix (PWM), which we call PWM2Vec, and use to generate feature vectors from the spike protein sequences of these coronaviruses. While our embedding is inspired by the success of PWMs in biological applications such as determining protein function, or identifying transcription factor binding sites, we are the first (to the best of our knowledge) to use PWMs in the context of host classification from viral sequences to generate a fixed-length feature vector representation. The results on the real world data show that in using PWM2Vec, we are able to perform comparably well as compared to baseline models. We also measure the importance of different amino acids using information gain to show the amino acids which are important for predicting the host of a given coronavirus.
Efficient Analysis of COVID-19 Clinical Data using Machine Learning Models
Oct 18, 2021


Because of the rapid spread of COVID-19 to almost every part of the globe, huge volumes of data and case studies have been made available, providing researchers with a unique opportunity to find trends and make discoveries like never before, by leveraging such big data. This data is of many different varieties, and can be of different levels of veracity e.g., precise, imprecise, uncertain, and missing, making it challenging to extract important information from such data. Yet, efficient analyses of this continuously growing and evolving COVID-19 data is crucial to inform -- often in real-time -- the relevant measures needed for controlling, mitigating, and ultimately avoiding viral spread. Applying machine learning based algorithms to this big data is a natural approach to take to this aim, since they can quickly scale to such data, and extract the relevant information in the presence of variety and different levels of veracity. This is important for COVID-19, and for potential future pandemics in general. In this paper, we design a straightforward encoding of clinical data (on categorical attributes) into a fixed-length feature vector representation, and then propose a model that first performs efficient feature selection from such representation. We apply this approach on two clinical datasets of the COVID-19 patients and then apply different machine learning algorithms downstream for classification purposes. We show that with the efficient feature selection algorithm, we can achieve a prediction accuracy of more than 90\% in most cases. We also computed the importance of different attributes in the dataset using information gain. This can help the policy makers to focus on only certain attributes for the purposes of studying this disease rather than focusing on multiple random factors that may not be very informative to patient outcomes.