 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBoosting t-SNE Efficiency for Sequencing Data: Insights from Kernel Selection
Dec 17, 2025



Dimensionality reduction techniques are essential for visualizing and analyzing high-dimensional biological sequencing data. t-distributed Stochastic Neighbor Embedding (t-SNE) is widely used for this purpose, traditionally employing the Gaussian kernel to compute pairwise similarities. However, the Gaussian kernel's lack of data-dependence and computational overhead limit its scalability and effectiveness for categorical biological sequences. Recent work proposed the isolation kernel as an alternative, yet it may not optimally capture sequence similarities. In this study, we comprehensively evaluate nine different kernel functions for t-SNE applied to molecular sequences, using three embedding methods: One-Hot Encoding, Spike2Vec, and minimizers. Through both subjective visualization and objective metrics (including neighborhood preservation scores), we demonstrate that the cosine similarity kernel in general outperforms other kernels, including Gaussian and isolation kernels, achieving superior runtime efficiency and better preservation of pairwise distances in low-dimensional space. We further validate our findings through extensive classification and clustering experiments across six diverse biological datasets (Spike7k, Host, ShortRead, Rabies, Genome, and Breast Cancer), employing multiple machine learning algorithms and evaluation metrics. Our results show that kernel selection significantly impacts not only visualization quality but also downstream analytical tasks, with the cosine similarity kernel providing the most robust performance across different data types and embedding strategies, making it particularly suitable for large-scale biological sequence analysis.
Computing Gram Matrix for SMILES Strings using RDKFingerprint and Sinkhorn-Knopp Algorithm
Dec 19, 2024In molecular structure data, SMILES (Simplified Molecular Input Line Entry System) strings are used to analyze molecular structure design. Numerical feature representation of SMILES strings is a challenging task. This work proposes a kernel-based approach for encoding and analyzing molecular structures from SMILES strings. The proposed approach involves computing a kernel matrix using the Sinkhorn-Knopp algorithm while using kernel principal component analysis (PCA) for dimensionality reduction. The resulting low-dimensional embeddings are then used for classification and regression analysis. The kernel matrix is computed by converting the SMILES strings into molecular structures using the Morgan Fingerprint, which computes a fingerprint for each molecule. The distance matrix is computed using the pairwise kernels function. The Sinkhorn-Knopp algorithm is used to compute the final kernel matrix that satisfies the constraints of a probability distribution. This is achieved by iteratively adjusting the kernel matrix until the marginal distributions of the rows and columns match the desired marginal distributions. We provided a comprehensive empirical analysis of the proposed kernel method to evaluate its goodness with greater depth. The suggested method is assessed for drug subcategory prediction (classification task) and solubility AlogPS ``Aqueous solubility and Octanol/Water partition coefficient" (regression task) using the benchmark SMILES string dataset. The outcomes show the proposed method outperforms several baseline methods in terms of supervised analysis and has potential uses in molecular design and drug discovery. Overall, the suggested method is a promising avenue for kernel methods-based molecular structure analysis and design.
EPIC: Enhancing Privacy through Iterative Collaboration
Nov 07, 2024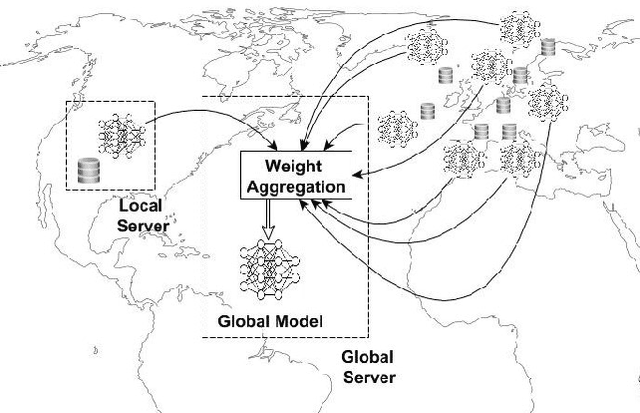
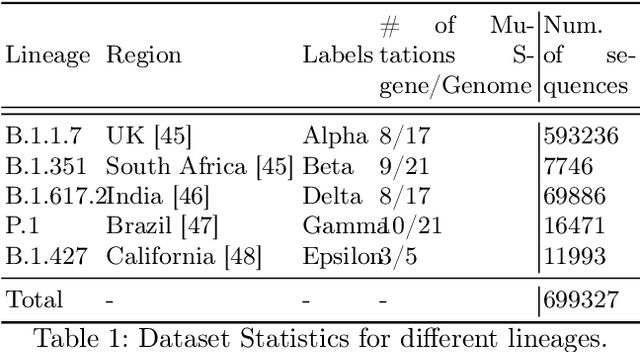
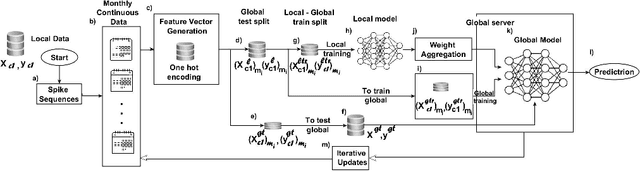
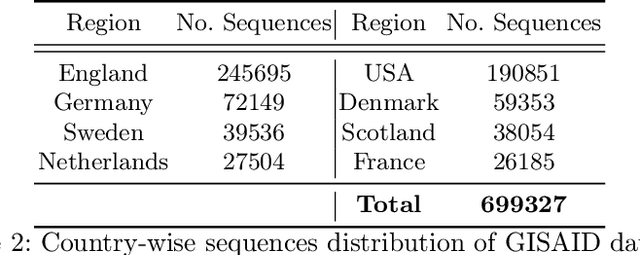
Advancements in genomics technology lead to a rising volume of viral (e.g., SARS-CoV-2) sequence data, resulting in increased usage of machine learning (ML) in bioinformatics. Traditional ML techniques require centralized data collection and processing, posing challenges in realistic healthcare scenarios. Additionally, privacy, ownership, and stringent regulation issues exist when pooling medical data into centralized storage to train a powerful deep learning (DL) model. The Federated learning (FL) approach overcomes such issues by setting up a central aggregator server and a shared global model. It also facilitates data privacy by extracting knowledge while keeping the actual data private. This work proposes a cutting-edge Privacy enhancement through Iterative Collaboration (EPIC) architecture. The network is divided and distributed between local and centralized servers. We demonstrate the EPIC approach to resolve a supervised classification problem to estimate SARS-CoV-2 genomic sequence data lineage without explicitly transferring raw sequence data. We aim to create a universal decentralized optimization framework that allows various data holders to work together and converge to a single predictive model. The findings demonstrate that privacy-preserving strategies can be successfully used with aggregation approaches without materially altering the degree of learning convergence. Finally, we highlight a few potential issues and prospects for study in FL-based approaches to healthcare applications.
DWFL: Enhancing Federated Learning through Dynamic Weighted Averaging
Nov 07, 2024Federated Learning (FL) is a distributed learning technique that maintains data privacy by providing a decentralized training method for machine learning models using distributed big data. This promising Federated Learning approach has also gained popularity in bioinformatics, where the privacy of biomedical data holds immense importance, especially when patient data is involved. Despite the successful implementation of Federated learning in biological sequence analysis, rigorous consideration is still required to improve accuracy in a way that data privacy should not be compromised. Additionally, the optimal integration of federated learning, especially in protein sequence analysis, has not been fully explored. We propose a deep feed-forward neural network-based enhanced federated learning method for protein sequence classification to overcome these challenges. Our method introduces novel enhancements to improve classification accuracy. We introduce dynamic weighted federated learning (DWFL) which is a federated learning-based approach, where local model weights are adjusted using weighted averaging based on their performance metrics. By assigning higher weights to well-performing models, we aim to create a more potent initial global model for the federated learning process, leading to improved accuracy. We conduct experiments using real-world protein sequence datasets to assess the effectiveness of DWFL. The results obtained using our proposed approach demonstrate significant improvements in model accuracy, making federated learning a preferred, more robust, and privacy-preserving approach for collaborative machine-learning tasks.
MIK: Modified Isolation Kernel for Biological Sequence Visualization, Classification, and Clustering
Oct 21, 2024



The t-Distributed Stochastic Neighbor Embedding (t-SNE) has emerged as a popular dimensionality reduction technique for visualizing high-dimensional data. It computes pairwise similarities between data points by default using an RBF kernel and random initialization (in low-dimensional space), which successfully captures the overall structure but may struggle to preserve the local structure efficiently. This research proposes a novel approach called the Modified Isolation Kernel (MIK) as an alternative to the Gaussian kernel, which is built upon the concept of the Isolation Kernel. MIK uses adaptive density estimation to capture local structures more accurately and integrates robustness measures. It also assigns higher similarity values to nearby points and lower values to distant points. Comparative research using the normal Gaussian kernel, the isolation kernel, and several initialization techniques, including random, PCA, and random walk initializations, are used to assess the proposed approach (MIK). Additionally, we compare the computational efficiency of all $3$ kernels with $3$ different initialization methods. Our experimental results demonstrate several advantages of the proposed kernel (MIK) and initialization method selection. It exhibits improved preservation of the local and global structure and enables better visualization of clusters and subclusters in the embedded space. These findings contribute to advancing dimensionality reduction techniques and provide researchers and practitioners with an effective tool for data exploration, visualization, and analysis in various domains.
Position Specific Scoring Is All You Need? Revisiting Protein Sequence Classification Tasks
Oct 16, 2024


Understanding the structural and functional characteristics of proteins are crucial for developing preventative and curative strategies that impact fields from drug discovery to policy development. An important and popular technique for examining how amino acids make up these characteristics of the protein sequences with position-specific scoring (PSS). While the string kernel is crucial in natural language processing (NLP), it is unclear if string kernels can extract biologically meaningful information from protein sequences, despite the fact that they have been shown to be effective in the general sequence analysis tasks. In this work, we propose a weighted PSS kernel matrix (or W-PSSKM), that combines a PSS representation of protein sequences, which encodes the frequency information of each amino acid in a sequence, with the notion of the string kernel. This results in a novel kernel function that outperforms many other approaches for protein sequence classification. We perform extensive experimentation to evaluate the proposed method. Our findings demonstrate that the W-PSSKM significantly outperforms existing baselines and state-of-the-art methods and achieves up to 45.1\% improvement in classification accuracy.
DANCE: Deep Learning-Assisted Analysis of Protein Sequences Using Chaos Enhanced Kaleidoscopic Images
Sep 10, 2024Cancer is a complex disease characterized by uncontrolled cell growth. T cell receptors (TCRs), crucial proteins in the immune system, play a key role in recognizing antigens, including those associated with cancer. Recent advancements in sequencing technologies have facilitated comprehensive profiling of TCR repertoires, uncovering TCRs with potent anti-cancer activity and enabling TCR-based immunotherapies. However, analyzing these intricate biomolecules necessitates efficient representations that capture their structural and functional information. T-cell protein sequences pose unique challenges due to their relatively smaller lengths compared to other biomolecules. An image-based representation approach becomes a preferred choice for efficient embeddings, allowing for the preservation of essential details and enabling comprehensive analysis of T-cell protein sequences. In this paper, we propose to generate images from the protein sequences using the idea of Chaos Game Representation (CGR) using the Kaleidoscopic images approach. This Deep Learning Assisted Analysis of Protein Sequences Using Chaos Enhanced Kaleidoscopic Images (called DANCE) provides a unique way to visualize protein sequences by recursively applying chaos game rules around a central seed point. we perform the classification of the T cell receptors (TCRs) protein sequences in terms of their respective target cancer cells, as TCRs are known for their immune response against cancer disease. The TCR sequences are converted into images using the DANCE method. We employ deep-learning vision models to perform the classification to obtain insights into the relationship between the visual patterns observed in the generated kaleidoscopic images and the underlying protein properties. By combining CGR-based image generation with deep learning classification, this study opens novel possibilities in the protein analysis domain.
Nearest Neighbor CCP-Based Molecular Sequence Analysis
Sep 07, 2024



Molecular sequence analysis is crucial for comprehending several biological processes, including protein-protein interactions, functional annotation, and disease classification. The large number of sequences and the inherently complicated nature of protein structures make it challenging to analyze such data. Finding patterns and enhancing subsequent research requires the use of dimensionality reduction and feature selection approaches. Recently, a method called Correlated Clustering and Projection (CCP) has been proposed as an effective method for biological sequencing data. The CCP technique is still costly to compute even though it is effective for sequence visualization. Furthermore, its utility for classifying molecular sequences is still uncertain. To solve these two problems, we present a Nearest Neighbor Correlated Clustering and Projection (CCP-NN)-based technique for efficiently preprocessing molecular sequence data. To group related molecular sequences and produce representative supersequences, CCP makes use of sequence-to-sequence correlations. As opposed to conventional methods, CCP doesn't rely on matrix diagonalization, therefore it can be applied to a range of machine-learning problems. We estimate the density map and compute the correlation using a nearest-neighbor search technique. We performed molecular sequence classification using CCP and CCP-NN representations to assess the efficacy of our proposed approach. Our findings show that CCP-NN considerably improves classification task accuracy as well as significantly outperforms CCP in terms of computational runtime.
Expanding Chemical Representation with k-mers and Fragment-based Fingerprints for Molecular Fingerprinting
Mar 28, 2024This study introduces a novel approach, combining substruct counting, $k$-mers, and Daylight-like fingerprints, to expand the representation of chemical structures in SMILES strings. The integrated method generates comprehensive molecular embeddings that enhance discriminative power and information content. Experimental evaluations demonstrate its superiority over traditional Morgan fingerprinting, MACCS, and Daylight fingerprint alone, improving chemoinformatics tasks such as drug classification. The proposed method offers a more informative representation of chemical structures, advancing molecular similarity analysis and facilitating applications in molecular design and drug discovery. It presents a promising avenue for molecular structure analysis and design, with significant potential for practical implementation.
* 12 Pages, 3 tables, Accepted at SimBig2023
A Universal Non-Parametric Approach For Improved Molecular Sequence Analysis
Feb 12, 2024

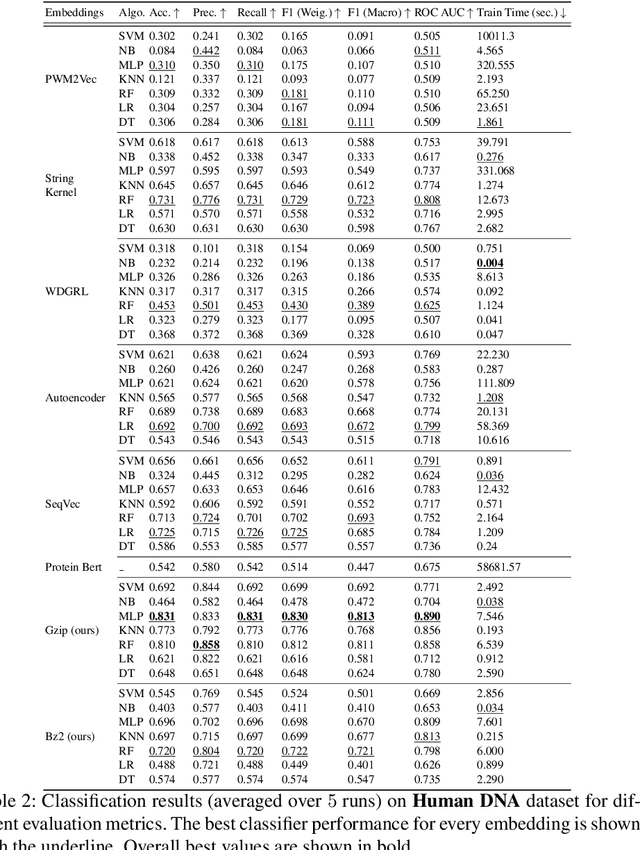
In the field of biological research, it is essential to comprehend the characteristics and functions of molecular sequences. The classification of molecular sequences has seen widespread use of neural network-based techniques. Despite their astounding accuracy, these models often require a substantial number of parameters and more data collection. In this work, we present a novel approach based on the compression-based Model, motivated from \cite{jiang2023low}, which combines the simplicity of basic compression algorithms like Gzip and Bz2, with Normalized Compression Distance (NCD) algorithm to achieve better performance on classification tasks without relying on handcrafted features or pre-trained models. Firstly, we compress the molecular sequence using well-known compression algorithms, such as Gzip and Bz2. By leveraging the latent structure encoded in compressed files, we compute the Normalized Compression Distance between each pair of molecular sequences, which is derived from the Kolmogorov complexity. This gives us a distance matrix, which is the input for generating a kernel matrix using a Gaussian kernel. Next, we employ kernel Principal Component Analysis (PCA) to get the vector representations for the corresponding molecular sequence, capturing important structural and functional information. The resulting vector representations provide an efficient yet effective solution for molecular sequence analysis and can be used in ML-based downstream tasks. The proposed approach eliminates the need for computationally intensive Deep Neural Networks (DNNs), with their large parameter counts and data requirements. Instead, it leverages a lightweight and universally accessible compression-based model.