 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeT Cell Receptor Protein Sequences and Sparse Coding: A Novel Approach to Cancer Classification
Apr 25, 2023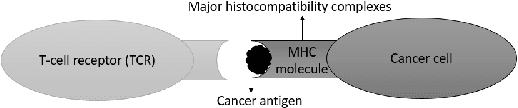
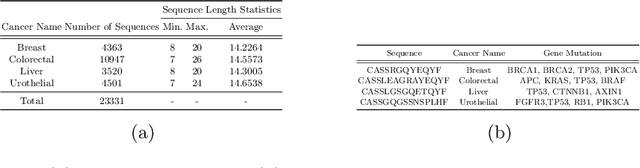
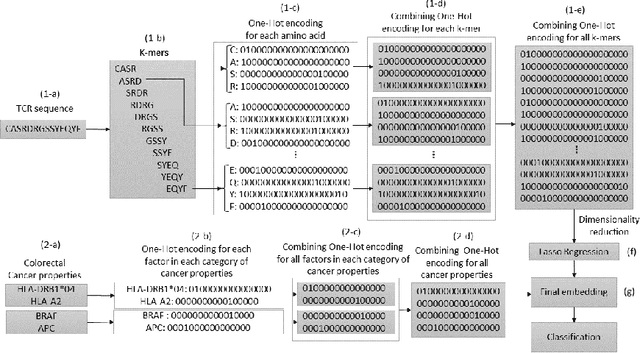
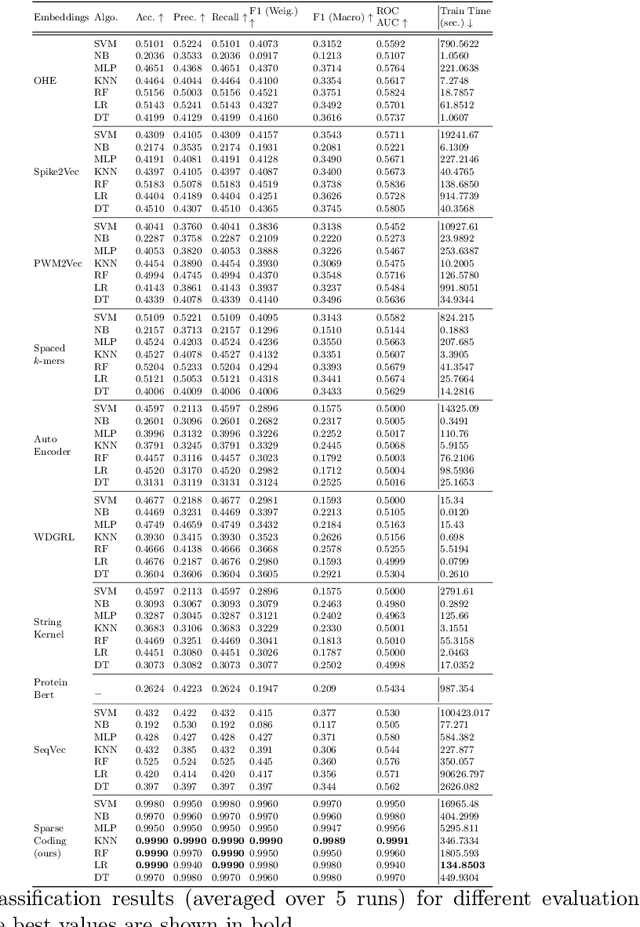
Cancer is a complex disease characterized by uncontrolled cell growth and proliferation. T cell receptors (TCRs) are essential proteins for the adaptive immune system, and their specific recognition of antigens plays a crucial role in the immune response against diseases, including cancer. The diversity and specificity of TCRs make them ideal for targeting cancer cells, and recent advancements in sequencing technologies have enabled the comprehensive profiling of TCR repertoires. This has led to the discovery of TCRs with potent anti-cancer activity and the development of TCR-based immunotherapies. In this study, we investigate the use of sparse coding for the multi-class classification of TCR protein sequences with cancer categories as target labels. Sparse coding is a popular technique in machine learning that enables the representation of data with a set of informative features and can capture complex relationships between amino acids and identify subtle patterns in the sequence that might be missed by low-dimensional methods. We first compute the k-mers from the TCR sequences and then apply sparse coding to capture the essential features of the data. To improve the predictive performance of the final embeddings, we integrate domain knowledge regarding different types of cancer properties. We then train different machine learning (linear and non-linear) classifiers on the embeddings of TCR sequences for the purpose of supervised analysis. Our proposed embedding method on a benchmark dataset of TCR sequences significantly outperforms the baselines in terms of predictive performance, achieving an accuracy of 99.8\%. Our study highlights the potential of sparse coding for the analysis of TCR protein sequences in cancer research and other related fields.
ViralVectors: Compact and Scalable Alignment-free Virome Feature Generation
Apr 07, 2023The amount of sequencing data for SARS-CoV-2 is several orders of magnitude larger than any virus. This will continue to grow geometrically for SARS-CoV-2, and other viruses, as many countries heavily finance genomic surveillance efforts. Hence, we need methods for processing large amounts of sequence data to allow for effective yet timely decision-making. Such data will come from heterogeneous sources: aligned, unaligned, or even unassembled raw nucleotide or amino acid sequencing reads pertaining to the whole genome or regions (e.g., spike) of interest. In this work, we propose \emph{ViralVectors}, a compact feature vector generation from virome sequencing data that allows effective downstream analysis. Such generation is based on \emph{minimizers}, a type of lightweight "signature" of a sequence, used traditionally in assembly and read mapping -- to our knowledge, the first use minimizers in this way. We validate our approach on different types of sequencing data: (a) 2.5M SARS-CoV-2 spike sequences (to show scalability); (b) 3K Coronaviridae spike sequences (to show robustness to more genomic variability); and (c) 4K raw WGS reads sets taken from nasal-swab PCR tests (to show the ability to process unassembled reads). Our results show that ViralVectors outperforms current benchmarks in most classification and clustering tasks.
Efficient Classification of SARS-CoV-2 Spike Sequences Using Federated Learning
Feb 17, 2023



This paper presents a federated learning (FL) approach to train an AI model for SARS-Cov-2 coronavirus variant identification. We analyze the SARS-CoV-2 spike sequences in a distributed way, without data sharing, to detect different variants of the rapidly mutating coronavirus. A vast amount of sequencing data of SARS-CoV-2 is available due to various genomic monitoring initiatives by several nations. However, privacy concerns involving patient health information and national public health conditions could hinder openly sharing this data. In this work, we propose a lightweight FL paradigm to cooperatively analyze the spike protein sequences of SARS-CoV-2 privately, using the locally stored data to train a prediction model from remote nodes. Our method maintains the confidentiality of local data (that could be stored in different locations) yet allows us to reliably detect and identify different known and unknown variants of the novel coronavirus SARS-CoV-2. We compare the performance of our approach on spike sequence data with the recently proposed state-of-the-art methods for classification from spike sequences. Using the proposed approach, we achieve an overall accuracy of $93\%$ on the coronavirus variant identification task. To the best of our knowledge, this is the first work in the federated learning paradigm for biological sequence analysis. Since the proposed model is distributed in nature, it could scale on ``Big Data'' easily. We plan to use this proof-of-concept to implement a privacy-preserving pandemic response strategy.
Robust Representation and Efficient Feature Selection Allows for Effective Clustering of SARS-CoV-2 Variants
Oct 18, 2021
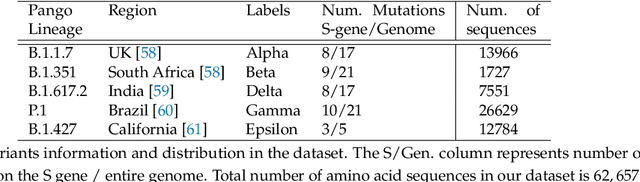
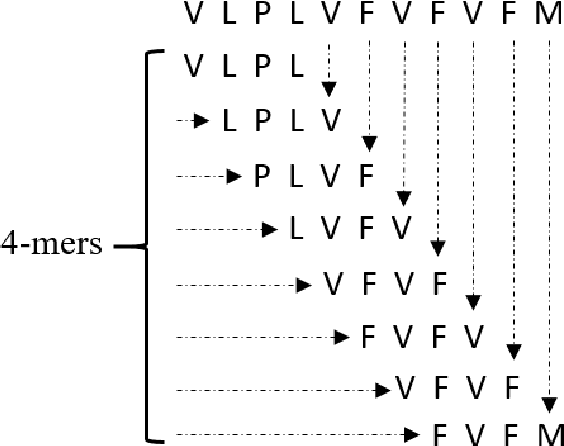
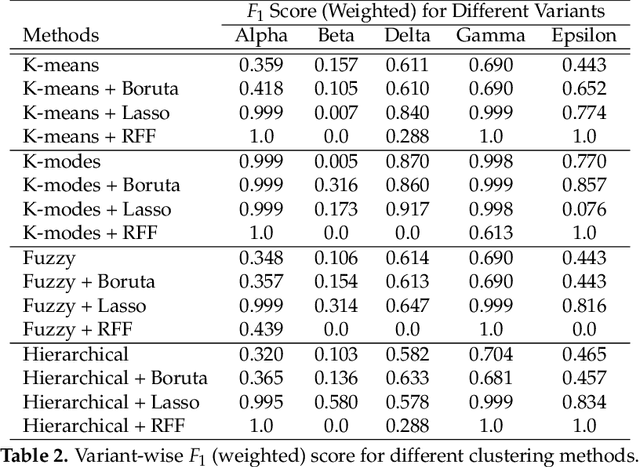
The widespread availability of large amounts of genomic data on the SARS-CoV-2 virus, as a result of the COVID-19 pandemic, has created an opportunity for researchers to analyze the disease at a level of detail unlike any virus before it. One one had, this will help biologists, policy makers and other authorities to make timely and appropriate decisions to control the spread of the coronavirus. On the other hand, such studies will help to more effectively deal with any possible future pandemic. Since the SARS-CoV-2 virus contains different variants, each of them having different mutations, performing any analysis on such data becomes a difficult task. It is well known that much of the variation in the SARS-CoV-2 genome happens disproportionately in the spike region of the genome sequence -- the relatively short region which codes for the spike protein(s). Hence, in this paper, we propose an approach to cluster spike protein sequences in order to study the behavior of different known variants that are increasing at very high rate throughout the world. We use a k-mers based approach to first generate a fixed-length feature vector representation for the spike sequences. We then show that with the appropriate feature selection, we can efficiently and effectively cluster the spike sequences based on the different variants. Using a publicly available set of SARS-CoV-2 spike sequences, we perform clustering of these sequences using both hard and soft clustering methods and show that with our feature selection methods, we can achieve higher F1 scores for the clusters.