 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRobust Representation and Efficient Feature Selection Allows for Effective Clustering of SARS-CoV-2 Variants
Paper and Code

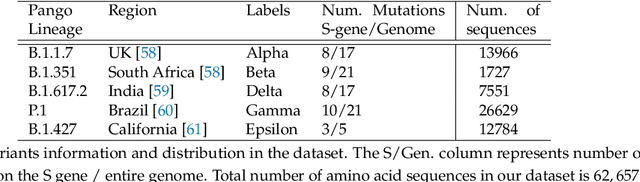
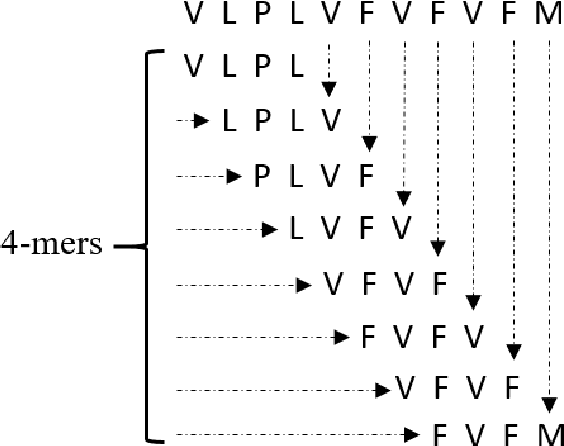
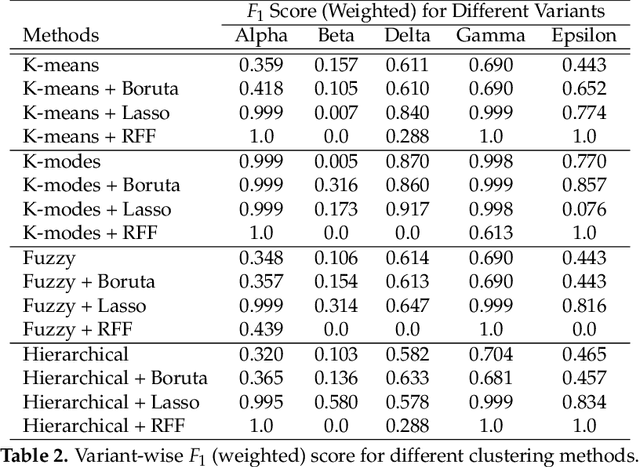
The widespread availability of large amounts of genomic data on the SARS-CoV-2 virus, as a result of the COVID-19 pandemic, has created an opportunity for researchers to analyze the disease at a level of detail unlike any virus before it. One one had, this will help biologists, policy makers and other authorities to make timely and appropriate decisions to control the spread of the coronavirus. On the other hand, such studies will help to more effectively deal with any possible future pandemic. Since the SARS-CoV-2 virus contains different variants, each of them having different mutations, performing any analysis on such data becomes a difficult task. It is well known that much of the variation in the SARS-CoV-2 genome happens disproportionately in the spike region of the genome sequence -- the relatively short region which codes for the spike protein(s). Hence, in this paper, we propose an approach to cluster spike protein sequences in order to study the behavior of different known variants that are increasing at very high rate throughout the world. We use a k-mers based approach to first generate a fixed-length feature vector representation for the spike sequences. We then show that with the appropriate feature selection, we can efficiently and effectively cluster the spike sequences based on the different variants. Using a publicly available set of SARS-CoV-2 spike sequences, we perform clustering of these sequences using both hard and soft clustering methods and show that with our feature selection methods, we can achieve higher F1 scores for the clusters.